Marginal log likelihood comparison of expression models
Table of Contents
Introduction
We previously used a goodness of fit test and out-of-sample (marginal) log likelihood to assess whether the data were adequately described by a given expression model. Here, we compute the in-sample marginal log likelihood, in order to make direct comparisons between expression models.
Setup
import anndata import numpy as np import os import pandas as pd import scanpy as sc import scipy.optimize as so import scipy.special as sp import scipy.stats as st import scmodes
import rpy2.robjects.packages import rpy2.robjects.pandas2ri rpy2.robjects.pandas2ri.activate() ashr = rpy2.robjects.packages.importr('ashr')
%matplotlib inline %config InlineBackend.figure_formats = set(['retina'])
import matplotlib.pyplot as plt plt.rcParams['figure.facecolor'] = 'w' plt.rcParams['font.family'] = 'Nimbus Sans'
Results
Data
Prepare the data in h5ad.
import functools as ft def read_chromium(sample): x = sc.read('/project2/mstephens/aksarkar/projects/singlecell-modes/data/negative-controls/svensson_chromium_control.h5ad') x = x[x.obs['sample'] == sample,x.var.filter(like='ERCC', axis='index').index] sc.pp.filter_genes(x, min_cells=1) x.var = x.var.reset_index() return x def read_dropseq(): x = sc.read('/project2/mstephens/aksarkar/projects/singlecell-modes/data/negative-controls/macosko_dropseq_control.h5ad') x = x[:,x.var.filter(like='ERCC', axis='index').index] sc.pp.filter_genes(x, min_cells=1) x.var = x.var.reset_index() return x def read_indrops(): x = sc.read('/project2/mstephens/aksarkar/projects/singlecell-modes/data/negative-controls/klein_indrops_control.h5ad') x = x[:,x.var.filter(like='ERCC', axis='index').index] sc.pp.filter_genes(x, min_cells=1) x.var = x.var.reset_index() return x def read_gemcode(): x = sc.read('/project2/mstephens/aksarkar/projects/singlecell-modes/data/negative-controls/zheng_gemcode_control.h5ad') x = x[:,x.var.filter(like='ERCC', axis='index').index] sc.pp.filter_genes(x, min_cells=1) x.var = x.var.reset_index() return x def _read_10x(k, return_df=False, min_detect=0.01, chunk=None, chunksize=500): x = scmodes.dataset.read_10x(f'/project2/mstephens/aksarkar/projects/singlecell-ideas/data/10xgenomics/{k}/filtered_matrices_mex/hg19/', return_adata=not return_df, return_df=return_df, min_detect=min_detect) x.var.columns = ['gene', 'name'] x.obs['size'] = x.X.sum(axis=1).A.ravel() if chunk is None: return x else: return x[:,chunk * chunksize:(chunk + 1) * chunksize] def _mix_10x(k1, k2, min_detect=0.01, return_y=False, chunk=None, chunksize=500): x1 = _read_10x(k1, min_detect=0) x2 = _read_10x(k2, min_detect=0) x = x1.concatenate(x2) sc.pp.filter_genes(x, min_cells=min_detect * x.shape[0]) x.obs['size'] = x.X.sum(axis=1).A.ravel() if chunk is None: return x else: return x[:,chunk * chunksize:(chunk + 1) * chunksize] def _cd8_cd19_mix(*args, **kwargs): return _mix_10x('cytotoxic_t', 'b_cells', *args, **kwargs) def _cyto_naive_mix(*args, **kwargs): return _mix_10x('cytotoxic_t', 'naive_t', *args, **kwargs) def read_ipsc(chunk=None, chunksize=500): x = anndata.read_h5ad('/project2/mstephens/aksarkar/projects/singlecell-ideas/data/ipsc/ipsc.h5ad') x.obs['size'] = x.X.sum(axis=1).A.ravel() if chunk is None: return x else: return x[:,chunk * chunksize:(chunk + 1) * chunksize] def read_liver(chunk=None, chunksize=500): x = anndata.read_h5ad('/project2/mstephens/aksarkar/projects/singlecell-ideas/data/human-cell-atlas/liver-caudate-lobe/liver-caudate-lobe.h5ad') x.obs['size'] = x.X.sum(axis=1).A.ravel() if chunk is None: return x else: return x[:,chunk * chunksize:(chunk + 1) * chunksize] def read_kidney(chunk=None, chunksize=500): x = anndata.read_h5ad('/project2/mstephens/aksarkar/projects/singlecell-ideas/data/human-cell-atlas/kidney/kidney.h5ad') x.obs['size'] = x.X.sum(axis=1).A.ravel() if chunk is None: return x else: return x[:,chunk * chunksize:(chunk + 1) * chunksize] def read_brain(chunk=None, chunksize=500): x = anndata.read_h5ad('/project2/mstephens/aksarkar/projects/singlecell-ideas/data/gtex-droncseq/gtex-droncseq.h5ad') sc.pp.filter_genes(x, min_counts=.01 * x.shape[0]) x.obs['size'] = x.X.sum(axis=1).A.ravel() if chunk is None: return x else: return x[:,chunk * chunksize:(chunk + 1) * chunksize] def read_retina(chunk=None, chunksize=500): x = anndata.read_h5ad('/project2/mstephens/aksarkar/projects/singlecell-ideas/data/human-cell-atlas/adult-retina/adult-retina.h5ad') query = x.obs['donor_organism.provenance.document_id'] == '427c0a62-9baf-42ab-a3a3-f48d10544280' y = x[query] sc.pp.filter_genes(y, min_cells=.01 * y.shape[0]) y.obs['size'] = y.X.sum(axis=1).A.ravel() if chunk is None: return y else: return y[:,chunk * chunksize:(chunk + 1) * chunksize] def read_pbmc_10k_v3(chunk=None, chunksize=500): x = anndata.read_h5ad('/scratch/midway2/aksarkar/modes/10k_pbmc_v3.h5ad') sc.pp.filter_genes(x, min_cells=0.01 * x.shape[0]) x.obs['size'] = x.X.sum(axis=1).A.ravel() if chunk is None: return x else: return x[:,chunk * chunksize:(chunk + 1) * chunksize] data = { 'dropseq': read_dropseq, 'indrops': read_indrops, 'chromium1': lambda: read_chromium('20311'), 'chromium2': lambda: read_chromium('20312'), 'gemcode': read_gemcode, 'cytotoxic_t': ft.partial(_read_10x, k='cytotoxic_t'), 'b_cells': ft.partial(_read_10x, k='b_cells'), 'ipsc': read_ipsc, 'cytotoxic_t-b_cells': _cd8_cd19_mix, 'cytotoxic_t-naive_t': _cyto_naive_mix, 'pbmc_10k_v3': read_pbmc_10k_v3, 'liver-caudate-lobe': read_liver, 'kidney': read_kidney, 'brain': read_brain, 'retina': read_retina, } chunksize = 500 chunks = { 'b_cells': 6417 // chunksize, 'brain': 11744 // chunksize, 'cytotoxic_t': 6530 // chunksize, 'cytotoxic_t-b_cells': 6647 // chunksize, 'cytotoxic_t-naive_t': 6246 // chunksize, 'ipsc': 9957 // chunksize, 'kidney': 15496 // chunksize, 'liver-caudate-lobe': 3181 // chunksize, 'pbmc_10k_v3': 12144 // chunksize, 'retina': 10047 // chunksize, } control = list(data.keys())[:5] non_control = list(data.keys())[5:]
Report the dimensions of each data set.
pd.DataFrame([data[k]().shape for k in data], columns=['num_samples', 'num_genes'], index=data.keys())
num_samples num_genes dropseq 84 81 indrops 953 103 chromium1 2000 88 chromium2 2000 88 gemcode 1015 91 cytotoxic_t 10209 6530 b_cells 10085 6417 ipsc 5597 9957 cytotoxic_t-b_cells 20294 6647 cytotoxic_t-naive_t 20688 6246 pbmc_10k_v3 11769 12144 liver-caudate-lobe 8856 16200 kidney 11233 15496 brain 14963 11744 retina 21285 10047
Estimate marginal log likelihood
Estimate the marginal likelihood for each data set, for each gene, for each family of expression models.
sbatch --partition=broadwl -n1 -c28 --exclusive --job-name=llik --time=30:00 -a 11-12 #!/bin/bash source activate scmodes python <<EOF <<imports>> import anndata import multiprocessing as mp import os <<data>> tasks = [(m, d) for m in ('point', 'gamma', 'point_gamma', 'unimodal', 'npmle') for d in data if not (m in ('unimodal', 'npmle') and d in chunks) and not (m == 'npmle' and d in control)] m, d = tasks[int(os.environ['SLURM_ARRAY_TASK_ID'])] with mp.Pool() as pool: x = data[d]() res = scmodes.benchmark.evaluate_llik(x, pool=pool, methods=[m]) res.to_csv(f'/scratch/midway2/aksarkar/modes/llik/{d}-{m}.txt.gz', compression='gzip', sep='\t') EOF
Fit non-parametric expression models serially for control data, to avoid memory issues.
for d in control: (scmodes.benchmark.evaluate_llik(data[d](), methods=['npmle'], max_grid_updates=20, tol=1e-5) .to_csv(f'/scratch/midway2/aksarkar/modes/llik/{d}-npmle.txt.gz', compression='gzip', sep='\t'))
Shard data sets to fit unimodal/non-parametric expression models within the
midway2 time/memory limits.
sbatch --partition=broadwl -n1 -c28 --exclusive --job-name=llik --time=4:00:00 -a 324-343 #!/bin/bash source activate scmodes python <<EOF <<imports>> import multiprocessing as mp import os <<data>> tasks = [(m, d, c) for m in ('unimodal', 'npmle') for d in chunks for c in range(chunks[d])] m, d, c = tasks[int(os.environ['SLURM_ARRAY_TASK_ID'])] kwargs = dict() if m == 'npmle': kwargs = {'max_grid_updates': 20, 'tol': 1e-4} with mp.Pool() as pool: x = data[d](chunk=c) res = scmodes.benchmark.evaluate_llik(x, s=x.obs['size'], pool=pool, methods=[m], **kwargs) res.to_csv(f'/scratch/midway2/aksarkar/modes/llik/{d}-{m}-{c}.txt.gz', compression='gzip', sep='\t') EOF
Move the results to permanent storage.
rsync -au /scratch/midway2/aksarkar/modes/llik/ /project2/mstephens/aksarkar/projects/singlecell-modes/data/llik/
Combine the sharded results.
for m in ('unimodal', 'npmle'): for k in chunks: if os.path.exists(f'/project2/mstephens/aksarkar/projects/singlecell-modes/data/llik/{k}-{m}-0.txt.gz'): (pd.concat([pd.read_csv(f'/project2/mstephens/aksarkar/projects/singlecell-modes/data/llik/{k}-{m}-{i}.txt.gz', index_col=0, sep='\t') for i in range(chunks[k])]) .to_csv(f'/project2/mstephens/aksarkar/projects/singlecell-modes/data/llik/{k}-{m}.txt.gz', sep='\t'))
Read the results.
llik = (pd.concat( { k: pd.concat([ pd.read_csv(f'/project2/mstephens/aksarkar/projects/singlecell-modes/data/llik/{k}-{m}.txt.gz', index_col=0, sep='\t') for m in ('point', 'gamma', 'point_gamma', 'unimodal', 'npmle') if os.path.exists(f'/project2/mstephens/aksarkar/projects/singlecell-modes/data/llik/{k}-{m}.txt.gz')]) for k in data }) .reset_index(level=0) .rename({'level_0': 'dataset'}, axis=1))
Report the number (fraction) of genes that have likelihood ratio greater than some threshold over some baseline. Ensure monotonicity post-hoc, to get around an issue where the unimodal model sometimes does worse than expected due to not optimizing over the granularity of the grid jointly with the mode.
def fix_llik(llik): llik['gamma'] = np.fmax(llik['gamma'], llik['point']) llik['point_gamma'] = np.fmax(llik['point_gamma'], llik['gamma']) llik['unimodal'] = np.fmax(llik['unimodal'], llik['gamma']) llik['npmle'] = np.fmax.reduce([llik['npmle'], llik['unimodal'], llik['point_gamma']]) return llik def compare(llik, baseline, thresh=10, agg=sum): labels = ['point', 'gamma', 'point_gamma', 'unimodal', 'npmle'] assert baseline in labels res = dict() for k, g in llik.groupby('dataset'): query = g.pivot_table(index='gene', columns='method', values='llik') query = fix_llik(query) res[k] = (query.sub(query[baseline], axis=0) > np.log(thresh)).agg(agg, axis=0) res = (pd.concat(res) .reset_index() .rename({'level_0': 'dataset'}, axis=1) .pivot_table(index='dataset', columns='method') [0]) del res[baseline] return res[[x for x in labels if x != baseline]]
Application to control data sets
For each control data set, for each expression model, report the fraction of genes better fit by a Gamma expression model than a point mass expression model.
compare(llik[llik['dataset'].isin(control)], baseline='point', thresh=10, agg=np.mean)
method gamma point_gamma unimodal npmle dataset chromium1 0.261364 0.295455 0.340909 0.352273 chromium2 0.704545 0.772727 0.738636 0.772727 dropseq 0.975309 0.975309 0.987654 0.987654 gemcode 0.615385 0.615385 0.659341 0.659341 indrops 0.087379 0.106796 0.155340 0.155340
For each control data set, for each expression model, report the number of genes which have improvement in marginal log likelihood above some threshold over the Gamma expression model.
compare(llik[llik['dataset'].isin(control)], baseline='gamma', thresh=10, agg=np.mean)
method point point_gamma unimodal npmle dataset chromium1 0.0 0.045455 0.215909 0.215909 chromium2 0.0 0.090909 0.193182 0.261364 dropseq 0.0 0.271605 0.691358 0.790123 gemcode 0.0 0.000000 0.637363 0.637363 indrops 0.0 0.000000 0.067961 0.067961
Find the genes where a point-Gamma model improved over a Gamma model.
temp = dict() for k, g in llik[llik['dataset'].isin(control)].groupby('dataset'): query = g.pivot_table(index='gene', columns='method', values='llik') query = fix_llik(query) temp[k] = query.loc[query['point_gamma'] > query['gamma'] + np.log(10)] query = (pd.concat(temp) .reset_index() .rename({'level_0': 'dataset'}, axis=1) [['dataset', 'gene', 'point', 'gamma', 'point_gamma', 'unimodal', 'npmle']])
Plot the distribution of likelihood ratios for unimodal vs. point-Gamma for those genes.
plt.clf() plt.gcf().set_size_inches(2.5, 2.5) plt.hist((query['unimodal'] - query['point_gamma']) / np.log(10), np.linspace(-5, 105, 23), color='0.7', density=False) plt.xlabel('log$_{10}$ likelihood ratio') plt.ylabel('Number of genes') plt.tight_layout()
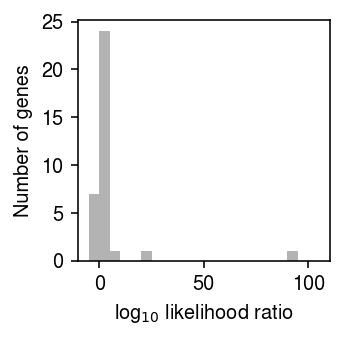
Of those genes, report the number of genes for which the likelihood ratio in favor of the unimodal model is not greater than one.
query[query['unimodal'] <= query['point_gamma']][['dataset', 'gene', 'point_gamma', 'unimodal', 'npmle']]
method dataset gene point_gamma unimodal npmle 7 chromium2 ERCC-00071 -2426.563183 -2426.598262 -2425.085059 8 chromium2 ERCC-00081 -17.679464 -21.457014 -17.679464 10 chromium2 ERCC-00116 -174.857339 -177.229879 -174.791905 11 chromium2 ERCC-00164 -48.993924 -53.541417 -48.993924 16 dropseq ERCC-00067 -60.613728 -61.697041 -59.606760 18 dropseq ERCC-00077 -142.917529 -143.542989 -142.729349 26 dropseq ERCC-00131 -341.742929 -347.518213 -337.359459
For each control data set, for each expression model, report the number of genes which have improvement in marginal log likelihood above some threshold over the unimodal expression model.
compare(llik[llik['dataset'].isin(control)], baseline='unimodal', thresh=10, agg=np.mean)
method point gamma point_gamma npmle dataset chromium1 0.0 0.0 0.000000 0.022727 chromium2 0.0 0.0 0.034091 0.034091 dropseq 0.0 0.0 0.012346 0.407407 gemcode 0.0 0.0 0.000000 0.120879 indrops 0.0 0.0 0.000000 0.000000
compare(llik[llik['dataset'].isin(control)], baseline='unimodal', thresh=50, agg=np.mean)
method point gamma point_gamma npmle dataset chromium1 0.0 0.0 0.000000 0.022727 chromium2 0.0 0.0 0.011364 0.011364 dropseq 0.0 0.0 0.012346 0.197531 gemcode 0.0 0.0 0.000000 0.010989 indrops 0.0 0.0 0.000000 0.000000
Look at the cases where NPMLE did better than unimodal.
temp = dict() for k, g in llik[llik['dataset'].isin(control)].groupby('dataset'): query = g.pivot_table(index='gene', columns='method', values='llik') query = fix_llik(query) temp[k] = query.loc[query['npmle'] > query['unimodal'] + np.log(10)] query = (pd.concat(temp) .reset_index() .rename({'level_0': 'dataset'}, axis=1) [['dataset', 'gene', 'point', 'gamma', 'point_gamma', 'unimodal', 'npmle']]) query
method dataset gene point gamma point_gamma \ 0 chromium1 ERCC-00074 -10994.555832 -10026.776094 -10026.776094 1 chromium1 ERCC-00130 -10649.942268 -9570.443230 -9565.932365 2 chromium2 ERCC-00081 -22.939272 -21.457014 -17.679464 3 chromium2 ERCC-00116 -179.364459 -177.229879 -174.857339 4 chromium2 ERCC-00164 -55.455464 -53.541417 -48.993924 5 dropseq ERCC-00002 -1266.423096 -611.402635 -611.394799 6 dropseq ERCC-00003 -1030.362549 -500.306576 -500.306517 7 dropseq ERCC-00004 -974.663055 -545.042159 -545.042159 8 dropseq ERCC-00019 -503.467112 -228.309470 -224.000070 9 dropseq ERCC-00025 -556.002790 -277.973395 -276.683859 10 dropseq ERCC-00034 -697.362242 -193.063000 -186.044807 11 dropseq ERCC-00039 -274.668122 -99.735102 -99.734779 12 dropseq ERCC-00042 -1153.137413 -483.932271 -483.732803 13 dropseq ERCC-00043 -1102.442375 -501.721756 -501.721753 14 dropseq ERCC-00044 -828.876427 -376.808649 -375.842974 15 dropseq ERCC-00053 -918.332827 -354.327829 -350.009082 16 dropseq ERCC-00060 -799.485748 -456.121055 -456.110088 17 dropseq ERCC-00062 -1155.604274 -370.017177 -369.687577 18 dropseq ERCC-00071 -856.315238 -360.975674 -357.071140 19 dropseq ERCC-00074 -1511.162109 -642.333522 -642.333522 20 dropseq ERCC-00076 -767.107849 -406.689904 -406.689904 21 dropseq ERCC-00079 -577.893220 -321.847139 -317.379547 22 dropseq ERCC-00084 -609.794518 -253.338088 -244.021041 23 dropseq ERCC-00085 -412.040273 -96.799237 -92.931414 24 dropseq ERCC-00092 -1082.852468 -421.866830 -420.327480 25 dropseq ERCC-00095 -683.153752 -381.605546 -381.578051 26 dropseq ERCC-00096 -1237.653809 -617.412762 -617.412762 27 dropseq ERCC-00108 -907.245140 -475.172703 -475.172647 28 dropseq ERCC-00112 -761.993153 -382.114154 -381.777423 29 dropseq ERCC-00113 -1074.117126 -538.694413 -538.694413 30 dropseq ERCC-00116 -694.544499 -367.351234 -364.166669 31 dropseq ERCC-00130 -1253.007568 -610.624357 -610.617778 32 dropseq ERCC-00131 -723.520929 -356.755781 -341.742929 33 dropseq ERCC-00144 -544.076984 -209.953994 -200.138600 34 dropseq ERCC-00145 -878.956902 -454.270630 -454.176155 35 dropseq ERCC-00160 -666.668557 -250.674782 -245.116842 36 dropseq ERCC-00165 -988.263237 -338.511813 -333.741251 37 dropseq ERCC-00168 -206.899650 -63.417678 -63.417678 38 gemcode ERCC-00009 -4510.433970 -4118.285506 -4118.285506 39 gemcode ERCC-00034 -1918.709922 -1755.789605 -1755.789560 40 gemcode ERCC-00035 -2716.800175 -2629.744168 -2629.744168 41 gemcode ERCC-00053 -2442.401288 -2282.002956 -2282.002774 42 gemcode ERCC-00060 -3244.700606 -3121.574336 -3121.571758 43 gemcode ERCC-00071 -3397.714914 -3153.035031 -3153.032709 44 gemcode ERCC-00078 -3001.456071 -2780.138679 -2780.138464 45 gemcode ERCC-00079 -3482.725608 -3123.857966 -3123.857966 46 gemcode ERCC-00085 -1168.949499 -1017.629741 -1017.629741 47 gemcode ERCC-00131 -2322.557758 -2232.505889 -2232.505889 48 gemcode ERCC-00160 -2675.612720 -2483.744290 -2483.744290 method unimodal npmle 0 -9842.350977 -9836.615070 1 -9517.260498 -9512.740669 2 -21.457014 -17.679464 3 -177.229879 -174.791905 4 -53.541417 -48.993924 5 -607.044232 -602.568171 6 -495.700019 -493.064685 7 -541.536046 -535.953133 8 -218.292515 -211.697691 9 -269.145382 -265.821108 10 -182.540192 -177.232823 11 -96.597446 -94.267453 12 -474.604140 -465.783799 13 -495.445359 -492.072696 14 -367.108223 -362.801161 15 -349.729527 -343.411482 16 -451.908588 -448.326980 17 -366.244318 -361.491025 18 -352.703587 -347.498260 19 -633.682726 -628.504607 20 -401.599442 -397.902139 21 -316.955326 -312.049294 22 -239.707689 -235.064641 23 -90.357506 -86.213971 24 -416.618139 -411.577139 25 -377.882827 -374.758270 26 -610.680517 -607.089068 27 -468.358776 -465.369989 28 -375.017848 -371.379920 29 -530.850285 -526.408639 30 -361.972045 -359.002267 31 -607.453443 -603.784909 32 -347.518213 -337.359459 33 -199.219463 -196.410539 34 -450.391125 -447.074294 35 -239.914256 -236.303790 36 -329.815130 -326.313666 37 -58.337531 -55.154366 38 -4077.611595 -4074.608323 39 -1642.184750 -1639.243826 40 -2524.676224 -2522.096733 41 -2144.102157 -2140.568698 42 -3029.049844 -3026.387906 43 -2987.354978 -2984.847676 44 -2637.040425 -2631.940091 45 -2924.518623 -2922.167038 46 -943.334301 -940.957160 47 -2141.876978 -2138.628185 48 -2348.491637 -2344.786146
Plot the results.
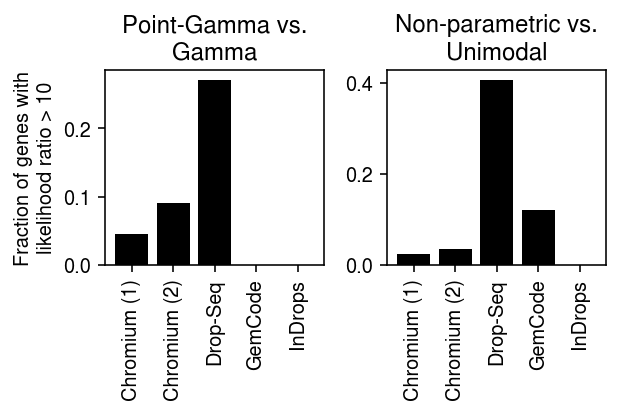
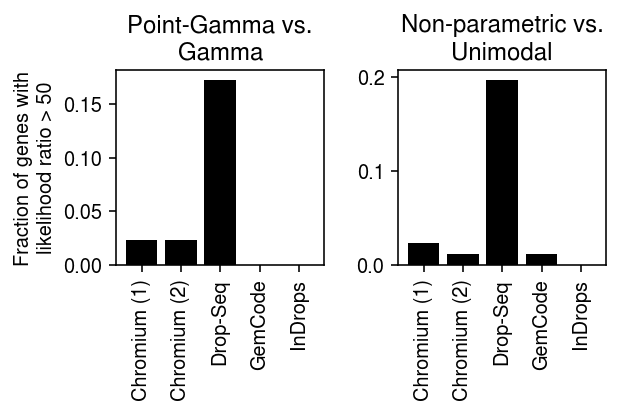
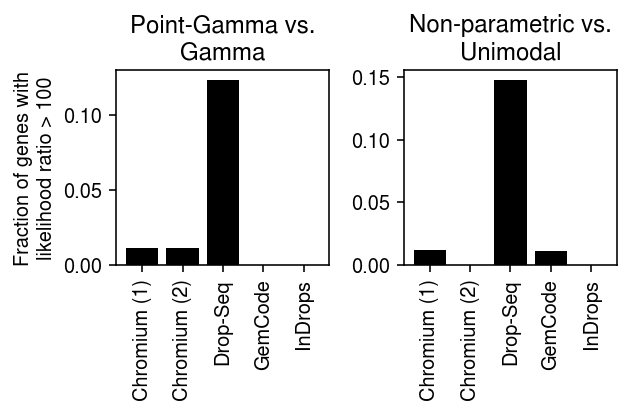
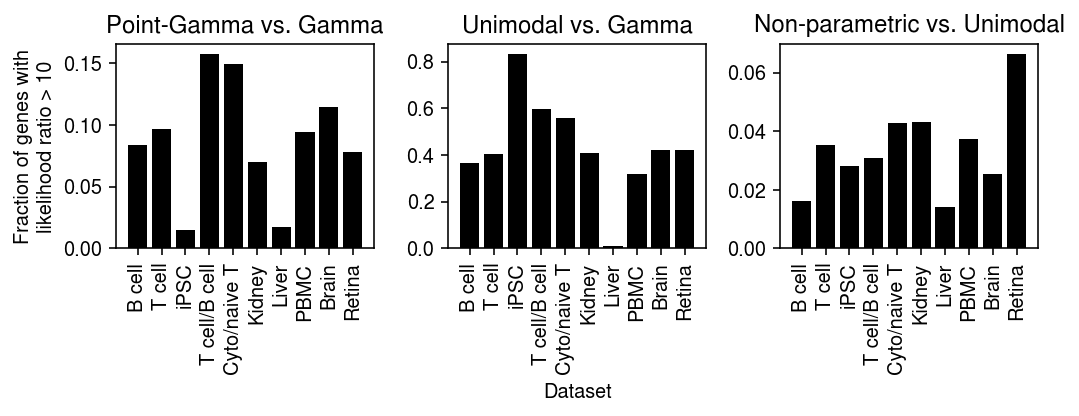
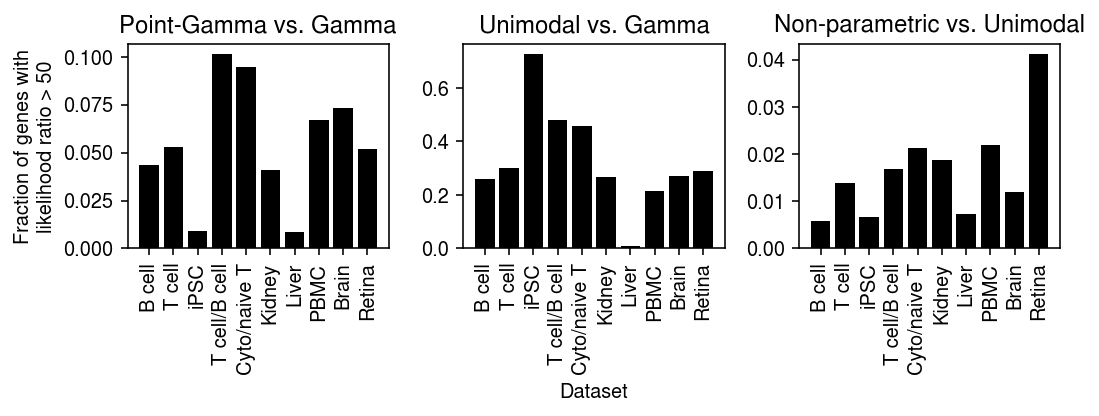
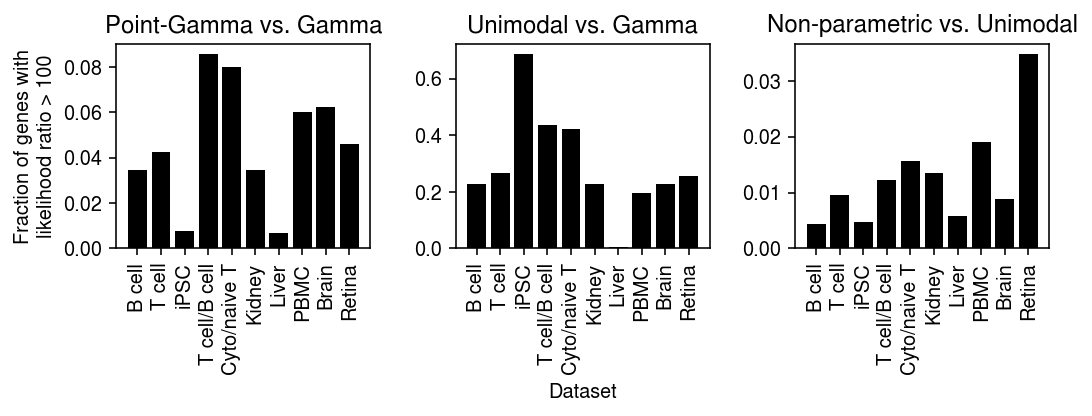
def _plot(llik, keys, labels, thresh=10): query1 = compare(llik, baseline='gamma', thresh=thresh, agg='mean') query2 = compare(llik, baseline='unimodal', thresh=thresh, agg='mean') plt.clf() fig, ax = plt.subplots(1, 2) fig.set_size_inches(5, 3) ax[0].bar(range(query1.shape[0]), query1.loc[keys, 'point_gamma'], color='k') ax[0].set_xticks(range(query1.shape[0])) ax[0].set_xticklabels(labels, rotation=90) ax[0].set_title('Point-Gamma vs.\nGamma') ax[0].set_ylabel(f'Fraction of genes with\nlikelihood ratio > {thresh}') ax[1].bar(range(query2.shape[0]), query2.loc[keys, 'npmle'], color='k') ax[1].set_xticks(range(query2.shape[0])) ax[1].set_xticklabels(labels, rotation=90) ax[1].set_title('Non-parametric vs.\nUnimodal') fig.tight_layout()
keys = ['chromium1', 'chromium2', 'dropseq', 'gemcode', 'indrops'] labels = ['Chromium (1)', 'Chromium (2)', 'Drop-Seq', 'GemCode', 'InDrops']
_plot_control(llik, thresh=10)
_plot_control(llik, thresh=50)
_plot_control(llik, thresh=100)
Application to biological data sets
Report the fraction of genes with strong evidence over a point mass expression model.
compare(llik[llik['dataset'].isin(non_control)], baseline='point', thresh=100, agg='mean')
method gamma point_gamma unimodal npmle dataset b_cells 0.637993 0.676017 0.707652 0.727131 brain 0.916042 0.942098 0.946185 0.954785 cytotoxic_t 0.635988 0.680092 0.709342 0.737519 cytotoxic_t-b_cells 0.848804 0.893937 0.901309 0.916052 cytotoxic_t-naive_t 0.761447 0.818764 0.835895 0.859110 ipsc 0.996284 0.997489 0.998895 0.998996 kidney 0.591443 0.605318 0.645199 0.654491 liver-caudate-lobe 0.120432 0.133642 0.127716 0.139136 pbmc_10k_v3 0.722744 0.737895 0.763093 0.771821 retina 0.488404 0.508112 0.569822 0.578282
The liver data shows little expression variation because the relative gene expression is low for almost all genes.
dat = data['liver-caudate-lobe']() fits = np.array([scmodes.ebpm.ebpm_point(dat.X[:,j].A.ravel(), dat.X.sum(axis=1).A.ravel()) for j in range(dat.shape[1])]).squeeze()
np.median(dat.X.sum(axis=1).A.ravel()) * np.percentile(np.exp(fits[:,0]), 99.7)
0.8378116504024844
This could mean some gene is dominating relative gene expression.
dat.var.iloc[np.argmax(fits[:,0])]
featurekey ENSG00000198804 featurename MT-CO1 featuretype protein_coding chromosome chrM featurestart 5904 featureend 7445 isgene True genus_species Homo sapiens n_cells 8856 Name: 17897, dtype: object
np.exp(np.max(fits[:,0]))
0.16776194506343123
Report the fraction of genes with strong evidence over a Gamma expression model.
compare(llik[llik['dataset'].isin(non_control)], baseline='gamma', thresh=100, agg='mean')
method point point_gamma unimodal npmle dataset b_cells 0.0 0.034751 0.228923 0.239676 brain 0.0 0.062415 0.227776 0.247701 cytotoxic_t 0.0 0.042726 0.265850 0.284533 cytotoxic_t-b_cells 0.0 0.085753 0.438092 0.455694 cytotoxic_t-naive_t 0.0 0.080051 0.421230 0.446846 ipsc 0.0 0.007733 0.687657 0.711861 kidney 0.0 0.034603 0.228800 0.252221 liver-caudate-lobe 0.0 0.006779 0.006481 0.012292 pbmc_10k_v3 0.0 0.060030 0.196146 0.207839 retina 0.0 0.046183 0.256893 0.278591
Report the fraction of genes with suggestive evidence over a Gamma expression model.
compare(llik[llik['dataset'].isin(non_control)], baseline='gamma', thresh=10, agg='mean')
method point point_gamma unimodal npmle dataset b_cells 0.0 0.083840 0.364812 0.391772 brain 0.0 0.114612 0.420044 0.459980 cytotoxic_t 0.0 0.096631 0.404747 0.449770 cytotoxic_t-b_cells 0.0 0.157665 0.597262 0.630961 cytotoxic_t-naive_t 0.0 0.149376 0.557157 0.603106 ipsc 0.0 0.014964 0.832580 0.854575 kidney 0.0 0.050336 0.365965 0.414107 liver-caudate-lobe 0.0 0.028395 0.019753 0.043519 pbmc_10k_v3 0.0 0.094779 0.316864 0.348403 retina 0.0 0.078133 0.421121 0.456057
Report the fraction of genes with strong evidence over a unimodal expression model.
compare(llik[llik['dataset'].isin(non_control)], baseline='unimodal', thresh=100, agg='mean')['npmle'].sort_values()
dataset b_cells 0.004363 ipsc 0.004821 liver-caudate-lobe 0.005736 brain 0.008941 cytotoxic_t 0.009648 cytotoxic_t-b_cells 0.012336 kidney 0.013481 cytotoxic_t-naive_t 0.015690 pbmc_10k_v3 0.019104 retina 0.034836 Name: npmle, dtype: float64
Plot the results.
keys = ['b_cells', 'cytotoxic_t', 'ipsc', 'cytotoxic_t-b_cells', 'cytotoxic_t-naive_t', 'kidney', 'liver-caudate-lobe', 'pbmc_10k_v3', 'brain', 'retina'] labels = ['B cell', 'T cell', 'iPSC', 'T cell/B cell', 'Cyto/naive T', 'Kidney', 'Liver', 'PBMC', 'Brain', 'Retina']
_plot(llik, thresh=10)
_plot(llik, thresh=50)
_plot(llik, thresh=100)
Drop-Seq spike-in gene
dat = data['dropseq']() x = dat[:,dat.var.iloc[:,0] == 'ERCC-00131'].X.A.ravel() s = dat.X.sum(axis=1).A.ravel() y = np.arange(x.max() + 1)
grid = np.linspace(0, (x / s).max(), 1000) pmf = dict() cdf = dict() gamma_res = scmodes.ebpm.ebpm_gamma(x, s) pmf['Gamma'] = np.array([scmodes.benchmark.gof._zig_pmf(k, size=s, log_mu=gamma_res[0], log_phi=-gamma_res[1]).mean() for k in y]) cdf['Gamma'] = st.gamma(a=np.exp(gamma_res[1]), scale=np.exp(gamma_res[0] - gamma_res[1])).cdf(grid) point_gamma_res = scmodes.ebpm.ebpm_point_gamma(x, s) cdf['ZIG'] = sp.expit(point_gamma_res[2]) + sp.expit(-point_gamma_res[2]) * st.gamma(a=np.exp(point_gamma_res[1]), scale=np.exp(point_gamma_res[0] - point_gamma_res[1])).cdf(grid) pmf['ZIG'] = np.array([scmodes.benchmark.gof._zig_pmf(k, size=s, log_mu=point_gamma_res[0], log_phi=-point_gamma_res[1], logodds=point_gamma_res[2]).mean() for k in y]) # Important: tune gridmult unimodal_res = ebpm_unimodal_tune(x, s) g = np.array(unimodal_res.rx2('fitted_g')) g = g[:,g[0] > 1e-8] a = np.fmin(g[1], g[2]) b = np.fmax(g[1], g[2]) comp_dens_conv = np.array([((st.gamma(a=k + 1, scale=1 / s.reshape(-1, 1)).cdf(b.reshape(1, -1)) - st.gamma(a=k + 1, scale=1 / s.reshape(-1, 1)).cdf(a.reshape(1, -1))) / np.outer(s, b - a)).mean(axis=0) for k in y]) comp_dens_conv[:,0] = st.poisson(mu=s.reshape(-1, 1) * b[0]).pmf(y).mean(axis=0) pmf['Unimodal'] = comp_dens_conv @ g[0] cdf['Unimodal'] = ashr.cdf_ash(unimodal_res, grid).rx2('y').ravel() npmle_res = scmodes.ebpm.ebpm_npmle(x, s, max_grid_updates=5, tol=1e-5) g = np.array(npmle_res.rx2('fitted_g')) g = g[:,g[0] > 1e-8] a = np.fmin(g[1], g[2]) b = np.fmax(g[1], g[2]) comp_dens_conv = np.array([((st.gamma(a=k + 1, scale=1 / s.reshape(-1, 1)).cdf(b.reshape(1, -1)) - st.gamma(a=k + 1, scale=1 / s.reshape(-1, 1)).cdf(a.reshape(1, -1))) / np.outer(s, b - a)).mean(axis=0) for k in y]) pmf['NPMLE'] = comp_dens_conv @ g[0] cdf['NPMLE'] = ashr.cdf_ash(npmle_res, grid).rx2('y').ravel()
1 - f4fd818b-e60b-4cc1-9c8b-69cfa67ba463
cm = plt.get_cmap('Paired') plt.clf() fig, ax = plt.subplots(2, 1) fig.set_size_inches(4.5, 4) ax[0].hist(x, bins=y, color='0.7', density=True) for i, k in enumerate(pmf): ax[0].plot(y + .5, pmf[k], lw=1, c=cm(i)) ax[0].set_xlabel('Number of molecules') ax[0].set_ylabel('Density') ax[0].set_title('ERCC-00131') for i, k in enumerate(pmf): ax[1].plot(grid, cdf[k], lw=1, c=cm(i), label=k) ax[1].legend(frameon=False) ax[1].set_xlabel('Latent gene expression') ax[1].set_ylabel('CDF') fig.tight_layout()
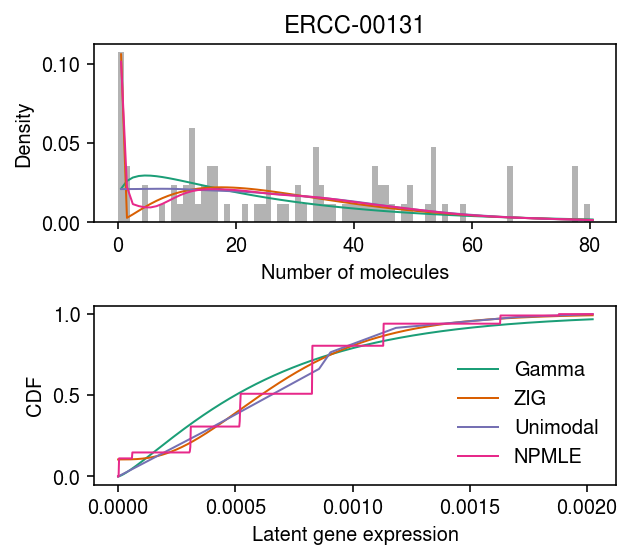
Issue in ash
Find all genes where a simpler expression model (point, Gamma, point-Gamma) did better than a unimodal expression model.
temp = dict() for k, g in llik[llik['dataset'].isin(non_control)].groupby('dataset'): query = g.pivot_table(index='gene', columns='method', values='llik') temp[k] = query.loc[(query.sub(query['unimodal'], axis=0)[['point', 'gamma', 'point_gamma']] > thresh).apply(any, axis=1)] query = (pd.concat(temp) .reset_index() .rename({'level_0': 'dataset'}, axis=1) [['dataset', 'gene', 'point', 'gamma', 'point_gamma', 'unimodal', 'npmle']]) query.shape
(3253, 7)
def ebpm_unimodal_tune(x, s): x = pd.Series(x) s = pd.Series(s) def _loss_gridmult(gridmult, mode, x, s): llik = ashr.ash_pois(x, s, mixcompdist='halfuniform', mode=mode, gridmult=gridmult, outputlevel='loglik', control=rpy2.robjects.ListVector({'tol.svd': 0})).rx2('loglik')[0] print(f'{mode:>9.6g} {gridmult:>9.6g} {llik:>9.6g}') return -llik def _loss_mode(mode, x, s): print(f'{"mode":>9s} {"gridmult":>9s} {"llik":>9s}') opt = so.minimize_scalar(_loss_gridmult, bracket=[1.1, 2], bounds=[1, 2], method='bounded', args=(mode, x, s), tol=1e-2) assert opt.success return opt.fun opt = so.minimize_scalar(_loss_mode, bracket=[0, (x / s).max()], bounds=[0, (x / s).max()], method='bounded', args=(x, s), tol=1e-5) mode = opt.x assert opt.success print(f'Converged to mode={mode:>9.6g}') opt = so.minimize_scalar(_loss_gridmult, bracket=[1.1, 2], bounds=[1, 2], method='bounded', args=(mode, x, s), tol=1e-2) assert opt.success gridmult = opt.x print(f'Converged to {gridmult:>9.6g}') res = ashr.ash_pois(x, s, mixcompdist='halfuniform', mode=mode, gridmult=gridmult, control=rpy2.robjects.ListVector({'tol.svd': 0})) return res
Report the fraction of genes for which NPMLE did better than the best of point mass, Gamma, and unimodal.
thresh = 10 temp = dict() for k, g in llik[llik['dataset'].isin(non_control)].groupby('dataset'): query = g.pivot_table(index='gene', columns='method', values='llik') baseline = query[['point', 'gamma', 'unimodal']].max(axis=1) temp[k] = (query['npmle'] > baseline + thresh).mean(axis=0) res = (pd.Series(temp) .reset_index() .rename({'index': 'dataset', 0: 'npmle'}, axis=1)) res
dataset npmle 0 b_cells 0.000156 1 brain 0.001448 2 cytotoxic_t 0.000766 3 cytotoxic_t-b_cells 0.003460 4 cytotoxic_t-naive_t 0.001441 5 ipsc 0.001908 6 kidney 0.003293 7 liver-caudate-lobe 0.000074 8 pbmc_10k_v3 0.010293 9 retina 0.013935
For each data set, for each expression model, report the number of genes which have improvement in marginal log likelihood above some threshold over the non-parametric expression model.
compare(llik[llik['dataset'].isin(non_control)], baseline='npmle', agg='mean')
method point gamma point_gamma unimodal dataset b_cells 0.106436 0.116254 0.116098 0.093657 brain 0.067439 0.103713 0.108396 0.112568 cytotoxic_t 0.096325 0.101378 0.101378 0.058499 cytotoxic_t-b_cells 0.113886 0.129081 0.130886 0.115390 cytotoxic_t-naive_t 0.124560 0.133045 0.134806 0.100865 ipsc 0.000000 0.000000 0.000000 0.000000 kidney 0.000000 0.017705 0.018140 0.018761 liver-caudate-lobe 0.000000 0.021605 0.021716 0.017582 pbmc_10k_v3 0.018281 0.034091 0.036397 0.037961 retina 0.000299 0.035334 0.036031 0.033841
For each control data set, for each expression model, report the number of genes which have improvement in marginal log likelihood above some threshold over the non-parametric expression model.
compare(llik[llik['dataset'].isin(control)], baseline='npmle')
method point gamma point_gamma unimodal dataset chromium1 0 1 0 2 chromium2 1 1 1 0 dropseq 0 0 0 0 gemcode 0 0 0 0 indrops 0 0 0 0
thresh = 10 temp = dict() for k, g in llik[llik['dataset'].isin(control)].groupby('dataset'): query = g.pivot_table(index='gene', columns='method', values='llik') temp[k] = query.loc[(query.sub(query['npmle'], axis=0) > thresh).apply(any, axis=1)] query = (pd.concat(temp) .reset_index() .rename({'level_0': 'dataset'}, axis=1) [['dataset', 'gene', 'point', 'gamma', 'point_gamma', 'unimodal', 'npmle']]) query
method dataset gene point gamma point_gamma \ 0 chromium1 ERCC-00054 -140.374338 -139.785657 -139.403474 1 chromium1 ERCC-00116 -110.987988 -103.145967 -108.102592 2 chromium2 ERCC-00142 -28.006585 -28.006585 -28.011618 method unimodal npmle 0 -139.360976 -149.364211 1 -102.207858 -115.402711 2 -148.213405 -43.356179
These cases can be explained by not starting the iterative refinement procedure with a fine enough grid.
temp = dict() for k, g in query.groupby('dataset'): dat = data[k]() for _, row in g.iterrows(): temp[(k, row['gene'])] = scmodes.ebpm.ebpm_npmle( dat[:,dat.var.iloc[:,0] == row['gene']].X.A.ravel(), dat.X.sum(axis=1).A.ravel(), K=1024, max_grid_updates=10, tol=1e-5, verbose=True).rx2('loglik')[0] pd.Series(temp).reset_index().rename({'level_0': 'dataset', 'level_1': 'gene', 0: 'npmle'}, axis=1)
dataset gene npmle 0 chromium1 ERCC-00054 -139.356572 1 chromium1 ERCC-00116 -102.119712 2 chromium2 ERCC-00142 -28.282124
Examples
Unimodal vs. Gamma
pivot = dict() for k, g in llik.groupby('dataset'): query = g.pivot_table(index='gene', columns='method', values='llik') query = fix_llik(query) pivot[k] = query pivot = pd.concat(pivot) query = (pivot .iloc[np.argsort(-pivot['unimodal'] + pivot['gamma'])] .reset_index() .rename({'level_0': 'dataset'}, axis=1)) query = query.loc[query['unimodal'] >= query['npmle'], ['dataset', 'gene', 'gamma', 'unimodal']] query.head(n=10)
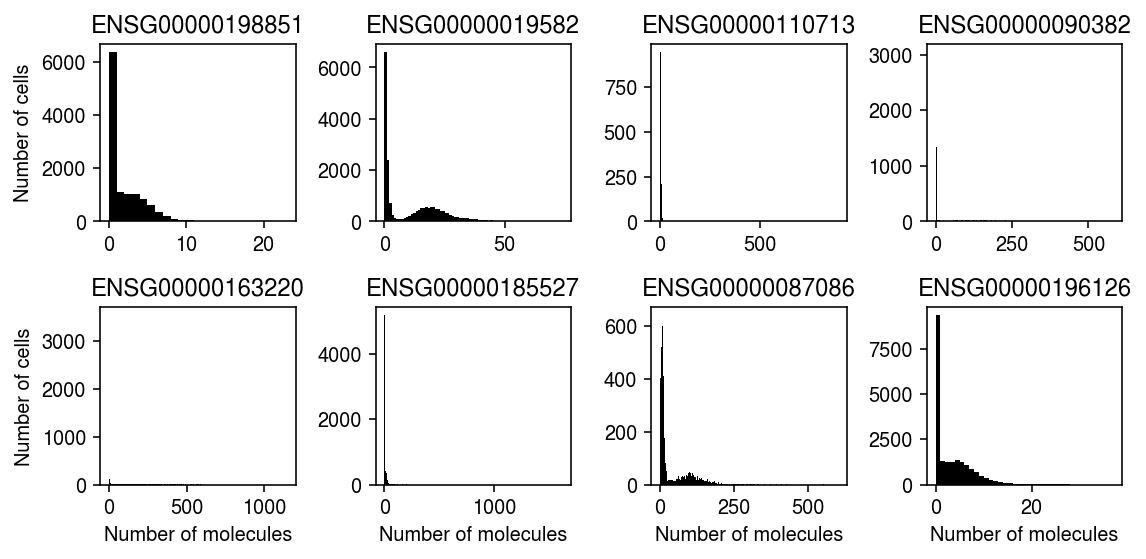
method dataset gene gamma unimodal 56 kidney ENSG00000196565 -5298.749426 -4178.292122 147 pbmc_10k_v3 ENSG00000163736 -6048.916822 -5388.534550 170 pbmc_10k_v3 ENSG00000180573 -6213.441429 -5615.144543 174 retina ENSG00000204091 -3643.764919 -3052.640395 188 pbmc_10k_v3 ENSG00000101856 -6772.157948 -6216.912371 202 kidney ENSG00000206172 -2534.658089 -2005.458099 220 pbmc_10k_v3 ENSG00000257764 -16933.243032 -16435.610729 230 brain VWF -1724.793605 -1253.016660 338 pbmc_10k_v3 ENSG00000211898 -8754.350377 -8427.946446 370 pbmc_10k_v3 ENSG00000211677 -8541.039933 -8244.471002
cm = plt.get_cmap('Dark2') plt.clf() fig, ax = plt.subplots(2, 4) fig.set_size_inches(8, 4) for a, (_, row) in zip(ax.ravel(), query.iterrows()): dat = data[row['level_0']]() x = dat[:,dat.var.iloc[:,0] == row['gene']].X.A.ravel() y = np.arange(x.max() + 1) a.hist(x, bins=y, color='k') a.set_title(row['gene']) for a in ax: a[0].set_ylabel('Number of cells') for a in ax.T: a[-1].set_xlabel('Number of molecules') ax[1,1].legend(frameon=False) fig.tight_layout()
dat = data['pbmc_10k_v3']() pmf = dict() cdf = dict() gene = 'ENSG00000163736' x = dat[:,dat.var['ensg'] == gene].X.A.ravel() s = dat.obs['size'] y = np.arange(x.max() + 2) lam = x / s grid = np.logspace(-11, -3, 1000) print(f'fitting {gene} (Gamma)') temp = scmodes.ebpm.ebpm_gamma(x, s, tol=1e-5, extrapolate=True) cdf['Gamma'] = st.gamma(a=np.exp(temp[1]), scale=np.exp(temp[0] - temp[1])).cdf(grid) pmf['Gamma'] = np.array([scmodes.benchmark.gof._zig_pmf(k, size=s, log_mu=temp[0], log_phi=-temp[1]).mean() for k in y]) print(f'fitting {gene} (Point-Gamma)') temp = scmodes.ebpm.ebpm_point_gamma(x, s, tol=1e-5, extrapolate=True) cdf['Point-Gamma'] = sp.expit(temp[2]) + sp.expit(-temp[2]) * st.gamma(a=np.exp(temp[1]), scale=np.exp(temp[0] - temp[1])).cdf(grid) pmf['Point-Gamma'] = np.array([scmodes.benchmark.gof._zig_pmf(k, size=s, log_mu=temp[0], log_phi=-temp[1], logodds=temp[2]).mean() for k in y]) print(f'fitting {gene} (Unimodal)') unimodal_res = scmodes.ebpm.ebpm_unimodal(x, s, mixcompdist='halfuniform') g = np.array(unimodal_res.rx2('fitted_g')) a = np.fmin(g[1], g[2]) b = np.fmax(g[1], g[2]) comp_dens_conv = np.array([((st.gamma(a=k + 1, scale=1 / s.values.reshape(-1, 1)).cdf(b.reshape(1, -1)) - st.gamma(a=k + 1, scale=1 / s.values.reshape(-1, 1)).cdf(a.reshape(1, -1))) / np.outer(s, b - a)).mean(axis=0) for k in y]) comp_dens_conv[:,0] = st.poisson(mu=s.values.reshape(-1, 1) * b[0]).pmf(y).mean(axis=0) pmf['Unimodal'] = comp_dens_conv @ g[0] cdf['Unimodal'] = ashr.cdf_ash(unimodal_res, grid).rx2('y').ravel()
cm = plt.get_cmap('Dark2') plt.clf() fig, ax = plt.subplots(2, 1) fig.set_size_inches(4, 4) for a, c in zip(ax, ['k', '0.7']): a.hist(x, bins=y, color=c) a.set_ylabel('Number of cells') for i, (k, ls) in enumerate(zip(pmf, ['-', (0, (3, 3)), '-'])): ax[1].plot(y + 0.5, x.shape[0] * pmf[k], lw=1, ls=ls, c=cm(i), label=k) ax[0].set_title(dat.var.loc[dat.var['ensg'] == gene, 'name'][0]) ax[1].set_xlabel('Number of molecules') ax[1].set_ylim(0, 10) ax[1].legend(frameon=False) fig.tight_layout()
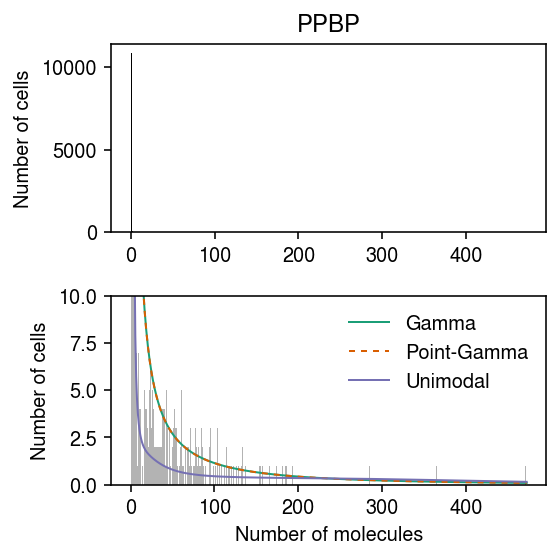
Examine why point-Gamma does not fit better than Gamma. First, check if the initialization is a problem.
dat = data['pbmc_10k_v3']() gene = 'ENSG00000163736' x = dat[:,dat.var['ensg'] == gene].X.A.ravel() s = dat.obs['size'].values.ravel() fit0 = pd.Series(scmodes.ebpm.ebpm_gamma(x, s, tol=1e-5, extrapolate=True), index=['log_mean', 'log_inv_disp', 'llik']) fit1 = pd.Series(scmodes.ebpm.ebpm_point_gamma(x, s, init=np.array([-sp.expit((x > 0).mean()), 1, s[x > 0].sum() / x[x > 0].sum()]), tol=1e-5, extrapolate=True), index=['log_mean', 'log_inv_disp', 'logodds', 'llik'])
pd.DataFrame({'gamma': fit0, 'point_gamma': fit1}).T
llik log_inv_disp log_mean logodds gamma -6048.895515 -4.267916 -7.292463 NaN point_gamma -6048.897291 -4.268733 -7.292503 -9.28471
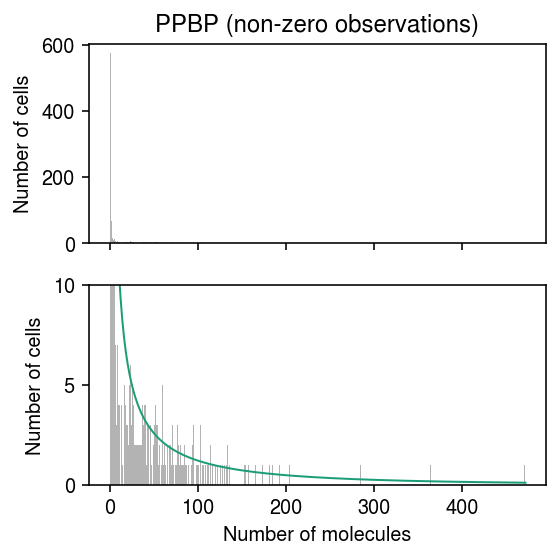
Plot the histogram restricting to entries greater than zero.
temp = scmodes.ebpm.ebpm_gamma(x[x > 0], s[x > 0], tol=1e-7, extrapolate=True) pmf = np.array([scmodes.benchmark.gof._zig_pmf(k, size=s, log_mu=temp[0], log_phi=-temp[1]).mean() for k in y]) plt.clf() fig, ax = plt.subplots(2, 1, sharex=True) fig.set_size_inches(4, 4) for a in ax: a.hist(x[x > 0], bins=y, color='0.7') a.set_ylabel('Number of cells') name = dat.var.loc[dat.var['ensg'] == gene, 'name'][0] ax[0].set_title(f'{name} (non-zero observations)') ax[1].plot(y + .5, (x > 0).sum() * pmf, lw=1, c=plt.get_cmap('Dark2')(0)) ax[1].set_ylim(0, 10) ax[1].set_yticks([0, 5, 10]) ax[1].set_xlabel('Number of molecules') fig.tight_layout()
Non-parametric vs. Gamma
Report the likelihood ratio for RPS4Y1.
temp = (query .iloc[np.argsort(-query['npmle'] + query['unimodal'])] .merge(dat.var, left_index=True, right_on='index') .query('name == "RPS4Y1"') [['unimodal', 'npmle']]) np.exp(temp['npmle'] - temp['unimodal'])
3911 5.912158e+65 dtype: float64
We previously used GOF to test whether the fitted point-Gamma expression model at each donor was adequate. Now, compare candidate models within each donor using log likelihood.
dat = data['ipsc']() x = dat[:,dat.var['name'] == 'RPS4Y1'].X.A.ravel() s = dat.obs['mol_hs'].values.ravel() fits = dict() for donor in dat.obs['chip_id'].unique(): idx = dat.obs['chip_id'] == donor for m in ('point', 'gamma', 'point_gamma'): print(f'fitting {donor} ({m})') fits[(donor, m)] = getattr(scmodes.ebpm, f'ebpm_{m}')(x[idx], s[idx])[-1] for m in ('unimodal', 'npmle'): print(f'fitting {donor} ({m})') fits[(donor, m)] = getattr(scmodes.ebpm, f'ebpm_{m}')(x[idx], s[idx]).rx2('loglik')[0]
rpsry1_res = (fix_llik(pd.DataFrame(fits, index=[0]).T .reset_index() .pivot_table(index='level_0', columns='level_1', values=0)) [['gamma', 'point_gamma', 'unimodal', 'npmle']])
rpsry1_res[rpsry1_res['unimodal'] > rpsry1_res['gamma'] + np.log(100)].shape[0]
28
rpsry1_res[rpsry1_res['unimodal'] > rpsry1_res['point_gamma'] + np.log(100)].shape[0]
11
rpsry1_res[rpsry1_res['npmle'] > rpsry1_res['unimodal'] + np.log(100)]
level_1 gamma point_gamma unimodal npmle level_0 NA18499 -317.457861 -290.457109 -309.357427 -289.500129 NA18522 -358.012379 -358.012379 -347.357464 -341.198174 NA19101 -337.264503 -337.264503 -327.732194 -317.848643 NA19152 -275.581828 -262.996097 -268.110564 -259.857938
query = pivot.loc['ipsc'] temp = (query .iloc[np.argsort(-query['npmle'] + query['unimodal'])] .merge(dat.var, left_index=True, right_on='index') .query('name == "SKP1"') [['unimodal', 'npmle']]) np.exp(temp['npmle'] - temp['unimodal'])
2717 3.115929 dtype: float64
x = dat[:,dat.var['name'] == 'SKP1'].X.A.ravel() s = dat.obs['mol_hs'].values.ravel() fits = dict() for donor in dat.obs['chip_id'].unique(): idx = dat.obs['chip_id'] == donor for m in ('point', 'gamma', 'point_gamma'): print(f'fitting {donor} ({m})') fits[(donor, m)] = getattr(scmodes.ebpm, f'ebpm_{m}')(x[idx], s[idx])[-1] for m in ('unimodal', 'npmle'): print(f'fitting {donor} ({m})') fits[(donor, m)] = getattr(scmodes.ebpm, f'ebpm_{m}')(x[idx], s[idx]).rx2('loglik')[0] skp1_res = (fix_llik(pd.DataFrame(fits, index=[0]).T .reset_index() .pivot_table(index='level_0', columns='level_1', values=0)) [['gamma', 'point_gamma', 'unimodal', 'npmle']])
skp1_res[skp1_res['point_gamma'] > skp1_res['gamma'] + np.log(100)].shape[0]
0
skp1_res[skp1_res['unimodal'] > skp1_res['gamma'] + np.log(100)].shape[0]
34
skp1_res[skp1_res['unimodal'] > skp1_res['point_gamma'] + np.log(100)].shape[0]
34
skp1_res[skp1_res['npmle'] > skp1_res['unimodal'] + np.log(100)]
level_1 gamma point_gamma unimodal npmle level_0 NA19116 -494.000236 -494.000236 -482.597503 -477.940124