Expression variation in human retina cells
Table of Contents
Introduction
Lukowski et al. 2019 generated a cell atlas of the human retina. Here, we study the prevalence of multi-modal expression variation in this data.
Setup
import anndata import numpy as np import pandas as pd import scanpy as sc import scipy.io as si import scipy.sparse as ss import scmodes import scmodes.benchmark.gof import scmodes.ebpm.sgd import torch
%matplotlib inline %config InlineBackend.figure_formats = set(['retina'])
import matplotlib.pyplot as plt plt.rcParams['figure.facecolor'] = 'w' plt.rcParams['font.family'] = 'Nimbus Sans'
Data
Read the data, and retain genes which have non-zero observations in at least 1% of cells.
prefix = '/project2/mstephens/aksarkar/projects/singlecell-ideas/data/human-cell-atlas/adult-retina/7f5e7a85-a45c-4535-90bf-0ef930a0040b.mtx' counts = si.mmread(f'{prefix}/matrix.mtx.gz').tocsr() # Important: the HCA metadata has a header, which breaks scmodes.dataset.read_10x samples = pd.read_csv(f'{prefix}/cells.tsv.gz', sep='\t') genes = pd.read_csv(f'{prefix}/genes.tsv.gz', sep='\t') x = anndata.AnnData(counts.T, obs=samples, var=genes) sc.pp.filter_genes(x, min_cells=.01 * x.shape[0]) x
AnnData object with n_obs × n_vars = 41350 × 11446 obs: 'cellkey', 'genes_detected', 'file_uuid', 'file_version', 'total_umis', 'emptydrops_is_cell', 'barcode', 'cell_suspension.provenance.document_id', 'specimen_from_organism.provenance.document_id', 'derived_organ_ontology', 'derived_organ_label', 'derived_organ_parts_ontology', 'derived_organ_parts_label', 'cell_suspension.genus_species.ontology', 'cell_suspension.genus_species.ontology_label', 'donor_organism.provenance.document_id', 'donor_organism.human_specific.ethnicity.ontology', 'donor_organism.human_specific.ethnicity.ontology_label', 'donor_organism.diseases.ontology', 'donor_organism.diseases.ontology_label', 'donor_organism.development_stage.ontology', 'donor_organism.development_stage.ontology_label', 'donor_organism.sex', 'donor_organism.is_living', 'specimen_from_organism.organ.ontology', 'specimen_from_organism.organ.ontology_label', 'specimen_from_organism.organ_parts.ontology', 'specimen_from_organism.organ_parts.ontology_label', 'library_preparation_protocol.provenance.document_id', 'library_preparation_protocol.input_nucleic_acid_molecule.ontology', 'library_preparation_protocol.input_nucleic_acid_molecule.ontology_label', 'library_preparation_protocol.library_construction_method.ontology', 'library_preparation_protocol.library_construction_method.ontology_label', 'library_preparation_protocol.end_bias', 'library_preparation_protocol.strand', 'project.provenance.document_id', 'project.project_core.project_short_name', 'project.project_core.project_title', 'analysis_protocol.provenance.document_id', 'dss_bundle_fqid', 'bundle_uuid', 'bundle_version', 'analysis_protocol.protocol_core.protocol_id', 'analysis_working_group_approval_status' var: 'featurekey', 'featurename', 'featuretype', 'chromosome', 'featurestart', 'featureend', 'isgene', 'genus_species', 'n_cells'
Write the h5ad out.
x.write('/project2/mstephens/aksarkar/projects/singlecell-ideas/data/human-cell-atlas/adult-retina/adult-retina.h5ad')
Report the number of samples per donor.
x.obs['donor_organism.provenance.document_id'].value_counts()
427c0a62-9baf-42ab-a3a3-f48d10544280 21285 b8049daa-7458-47bf-8ec2-3f5c56d2cb34 16164 66b7152c-8e94-412b-b0e4-f8fbf554df5a 3901 Name: donor_organism.provenance.document_id, dtype: int64
Read the h5ad.
x = anndata.read_h5ad('/project2/mstephens/aksarkar/projects/singlecell-ideas/data/human-cell-atlas/adult-retina/adult-retina.h5ad')
Results
Gamma assumption
Fit a Gamma distribution to expression variation at each gene within the donor with maximum number of cells assayed.
query = x.obs['donor_organism.provenance.document_id'] == '427c0a62-9baf-42ab-a3a3-f48d10544280'
res = scmodes.ebpm.sgd.ebpm_gamma(x[query].X, batch_size=64, max_epochs=20, verbose=True)
Test GOF at each gene.
s = x[query].X.sum(axis=1).A.ravel() gof_res = [] for j, (log_mu, neg_log_phi) in enumerate(np.vstack(res[:-1]).T): d, p = scmodes.benchmark.gof._gof(x=x[query,j].X.A.ravel(), cdf=scmodes.benchmark.gof._zig_cdf, pmf=scmodes.benchmark.gof._zig_pmf, size=s, log_mu=log_mu, log_phi=-neg_log_phi) gof_res.append([d, p]) gof_res = pd.DataFrame(gof_res, columns=['stat', 'p'], index=x.var['featurename']).reset_index() gof_res.to_csv('/project2/mstephens/aksarkar/projects/singlecell-modes/data/gof/adult-retina-gamma.txt.gz', sep='\t', compression='gzip')
Plot the histogram of GOF \(p\)-values.
plt.clf() plt.gcf().set_size_inches(2, 2) plt.hist(gof_res['p'], bins=np.linspace(0, 1, 11), color='0.7', density=True) plt.axhline(y=1, lw=1, ls=':', c='k') plt.xlim(0, 1) plt.xlabel('$p$-value') plt.ylabel('Density') plt.tight_layout()
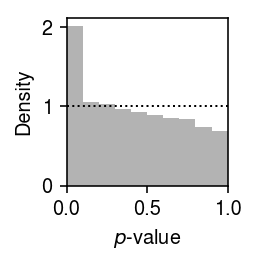
Report the fraction of genes which significantly depart from Gamma (Bonferroni-corrected \(p < 0.05\)).
sig = gof_res.loc[gof_res['p'] < 0.05 / gof_res.shape[0]] sig.shape[0] / gof_res.shape[0]
0.020094356106936922
Point-Gamma assumption
Fit a point-Gamma distribution to expression variation at each gene.
res = scmodes.ebpm.sgd.ebpm_point_gamma(x[query].X, max_epochs=20, verbose=True)
Test GOF at each gene.
s = x[query].X.sum(axis=1).A.ravel() result = [] for j, (log_mu, neg_log_phi, logodds) in enumerate(np.vstack(res[:3]).T): d, p = scmodes.benchmark.gof._gof(x=x[query,j].X.A.ravel(), cdf=scmodes.benchmark.gof._zig_cdf, pmf=scmodes.benchmark.gof._zig_pmf, size=s, log_mu=log_mu, log_phi=-neg_log_phi, logodds=logodds) result.append([d, p]) result = pd.DataFrame(result, columns=['stat', 'p'], index=x.var['featurename']).reset_index() result.to_csv('/project2/mstephens/aksarkar/projects/singlecell-modes/data/gof/retina-point-gamma.txt.gz', sep='\t', compression='gzip')
Plot the histogram of GOF \(p\)-values.
plt.clf() plt.gcf().set_size_inches(2, 2) plt.hist(result['p'], bins=np.linspace(0, 1, 11), color='0.7', density=True) plt.axhline(y=1, lw=1, ls=':', c='k') plt.xlim(0, 1) plt.xlabel('$p$-value') plt.ylabel('Density') plt.tight_layout()
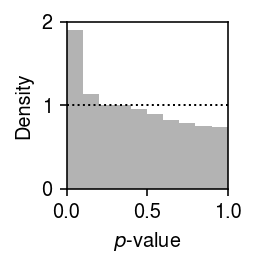
Report the fraction of genes which significantly depart from point-Gamma (Bonferroni-corrected \(p < 0.05\)).
sig = result.loc[result['p'] < 0.05 / result.shape[0]] sig.shape[0] / result.shape[0]
0.01712388607373755
Write out the significant genes.
x.obs['size'] = x.X.sum(axis=1) x[query,sig.index].write('/scratch/midway2/aksarkar/modes/unimodal-data/retina.h5ad')
Unimodal assumption
Fit a unimodal distribution to expression variation at each gene which departed significantly from a point-Gamma assumption.
sbatch --partition=broadwl -n1 -c28 --exclusive --time=12:00:00 --job-name=gof #!/bin/bash source activate scmodes python <<EOF import anndata import multiprocessing as mp import pandas as pd import scmodes with mp.Pool(maxtasksperchild=20) as pool: x = anndata.read('/scratch/midway2/aksarkar/modes/unimodal-data/retina.h5ad') res = scmodes.benchmark.evaluate_gof(pd.DataFrame(x.X.A), s=x.obs['size'], pool=pool, methods=['unimodal']) res.index = x.var['featurename'] res.to_csv(f'/project2/mstephens/aksarkar/projects/singlecell-modes/data/gof/retina-unimodal.txt.gz', compression='gzip', sep='\t') EOF
Read the results.
unimodal_res = pd.read_csv('/project2/mstephens/aksarkar/projects/singlecell-modes/data/gof/retina-unimodal.txt.gz', sep='\t', index_col=0)
Plot the histogram of GOF \(p\)-values.
plt.clf() plt.gcf().set_size_inches(2, 2) plt.hist(unimodal_res['p'], bins=np.linspace(0, 1, 11), color='0.7', density=True) plt.axhline(y=1, lw=1, ls=':', c='k') plt.xlim(0, 1) plt.xlabel('$p$-value') plt.ylabel('Density') plt.tight_layout()
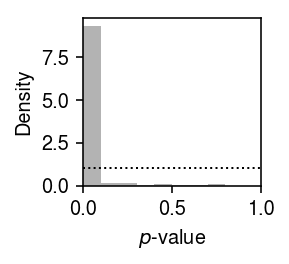
Report the number and fraction of genes which depart from a unimodal assumption.
sig = unimodal_res.loc[unimodal_res['p'] < .05 / unimodal_res.shape[0]] sig.shape[0], sig.shape[0] / x.shape[1]
(124, 0.010833478944609471)