Distribution deconvolution examples
Table of Contents
Introduction
We systematically compared different deconvolution methods on their ability to generalize to held out data. Here, we look at the data and fitted distributions for specific examples.
Setup
import functools as ft import multiprocessing as mp import numpy as np import pandas as pd import scanpy import scipy.stats as st import scipy.special as sp import scmodes import sklearn.model_selection as skms import rpy2.robjects.packages import rpy2.robjects.pandas2ri import rpy2.robjects.numpy2ri rpy2.robjects.pandas2ri.activate() rpy2.robjects.numpy2ri.activate() ashr = rpy2.robjects.packages.importr('ashr') descend = rpy2.robjects.packages.importr('descend')
%matplotlib inline %config InlineBackend.figure_formats = set(['retina'])
import colorcet import matplotlib.pyplot as plt plt.rcParams['figure.facecolor'] = 'w' plt.rcParams['font.family'] = 'Nimbus Sans'
Results
Test
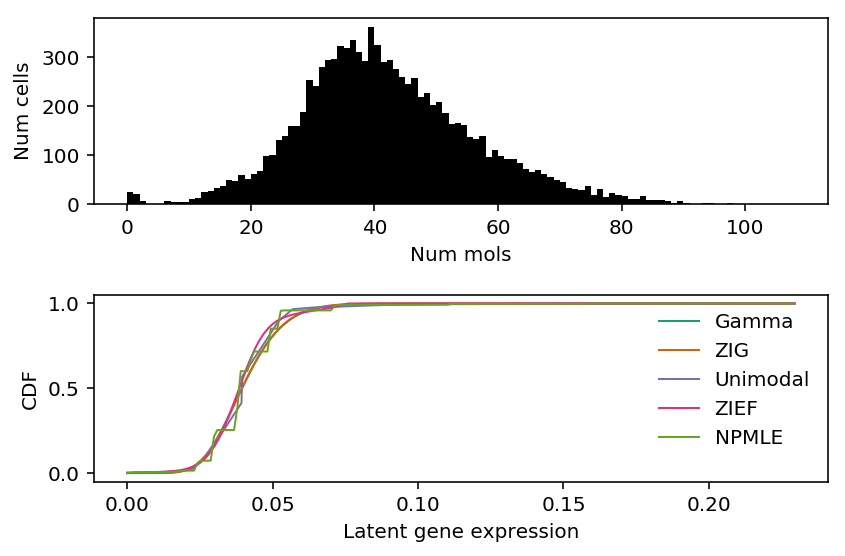
b_cells = scmodes.dataset.read_10x('/project2/mstephens/aksarkar/projects/singlecell-ideas/data/10xgenomics/b_cells/filtered_matrices_mex/hg19/', return_df=True) llik_diff = benchmark['b_cells']['npmle'] - benchmark['b_cells']['gamma'] query = llik_diff[llik_diff > 0].sort_values(ascending=False).head(n=1).index
x = b_cells[query].values.ravel() s = b_cells.sum(axis=1).values.ravel()
plt.clf() fig, ax = plt.subplots(2, 1) fig.set_size_inches(6, 4) ax[0].hist(x, bins=np.arange(x.max() + 1), color='k') ax[0].set_xlabel('Num mols') ax[0].set_ylabel('Num cells') for c, k in zip(plt.get_cmap('Dark2').colors, ['Gamma', 'ZIG', 'Unimodal', 'ZIEF', 'NPMLE']): ax[1].plot(*getattr(scmodes.deconvolve, f'fit_{k.lower()}')(x, s), color=c, lw=1, label=k) ax[1].set_xlabel('Latent gene expression') ax[1].set_ylabel('CDF') ax[1].legend(frameon=False) fig.tight_layout()
Metadata
Read gene metadata.
gene_info = pd.read_csv('/project2/mstephens/aksarkar/projects/singlecell-qtl/data/scqtl-genes.txt.gz', sep='\t', index_col=0)
Spike-in data
Read the results.
benchmark = { k: g.reset_index(level=0, drop=True) for k, g in (pd.read_csv('/project2/mstephens/aksarkar/projects/singlecell-modes/data/deconv-generalization/control-kl.txt.gz', sep='\t', index_col=[0, 1]) .groupby(level=0))}
We are primarily interested in cases where NPMLE does better than unimodal, because these could potentially be cases where zeros drive the difference in generalization performance.
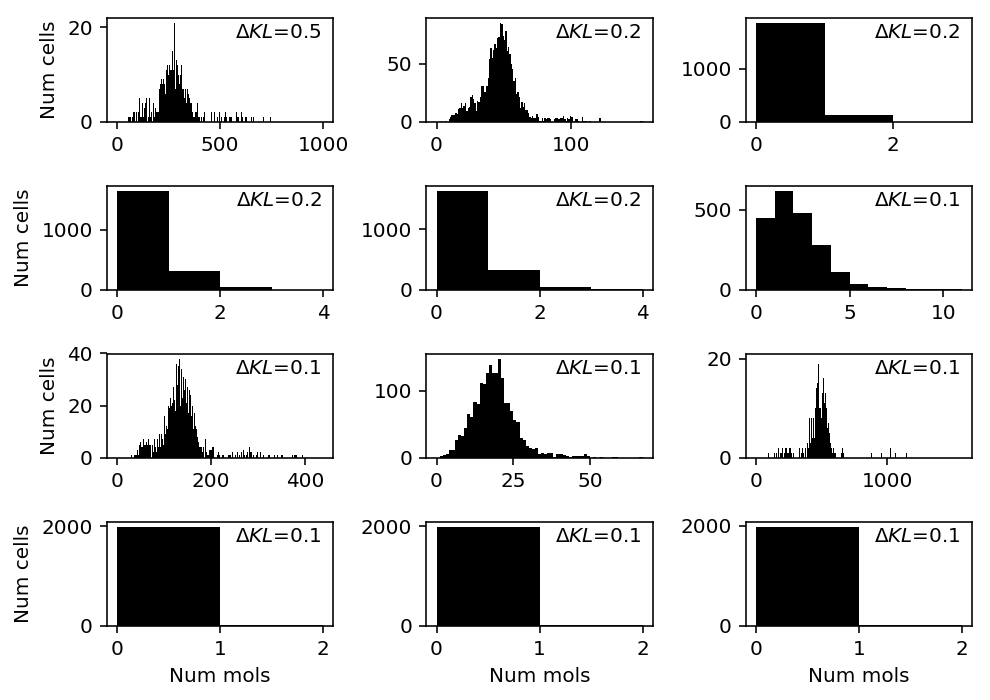
chromium1 = data['chromium1']() llik_diff = benchmark['chromium1']['npmle'] - benchmark['chromium1']['unimodal'] query = llik_diff[np.logical_and(chromium1.sum(axis=0) > 0, llik_diff > 0)].sort_values(ascending=False).head(n=12).index plt.clf() fig, ax = plt.subplots(4, 3) fig.set_size_inches(7, 5) for a, k in zip(ax.ravel(), query): a.hist(chromium1[:,k], bins=np.arange(chromium1[:,k].max() + 2), color='k') a.text(x=.95, y=.95, s=f"$\Delta KL$={llik_diff.loc[k]:.1f}", horizontalalignment='right', verticalalignment='top', transform=a.transAxes) for y in range(ax.shape[0]): ax[y][0].set_ylabel('Num cells') for x in range(ax.shape[1]): ax[-1][x].set_xlabel('Num mols') fig.tight_layout()
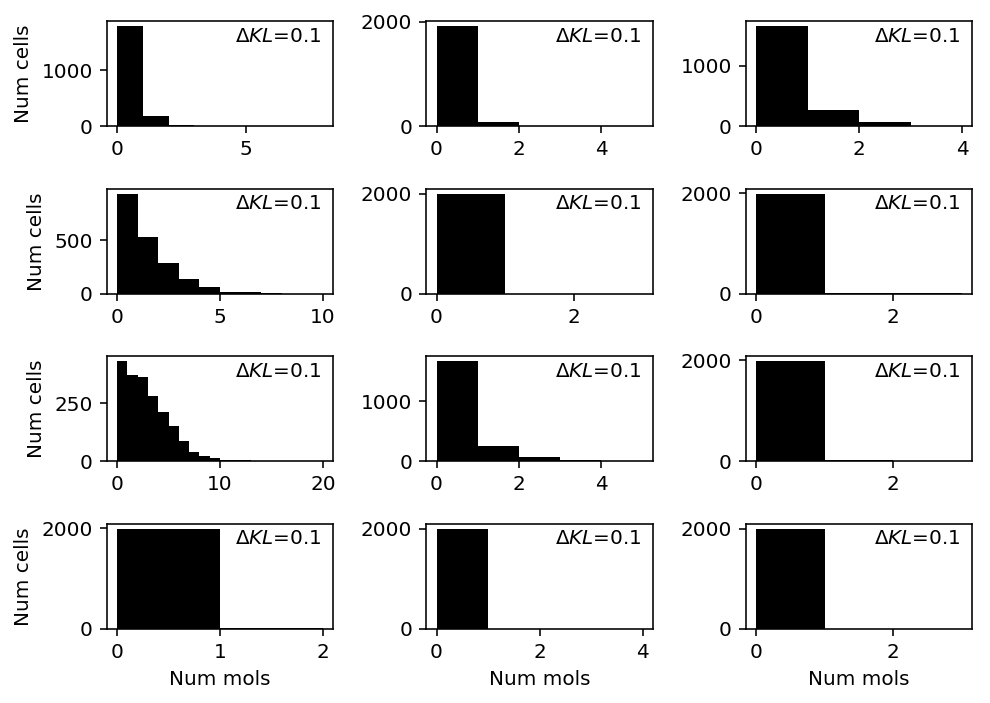
chromium2 = data['chromium2']() llik_diff = benchmark['chromium2']['npmle'] - benchmark['chromium2']['unimodal'] query = llik_diff[np.logical_and(chromium2.sum(axis=0) > 0, llik_diff > 0)].sort_values(ascending=False).head(n=12).index
plt.clf() fig, ax = plt.subplots(4, 3) fig.set_size_inches(7, 5) for a, k in zip(ax.ravel(), query): a.hist(chromium2[:,k], bins=np.arange(chromium2[:,k].max() + 2), color='k') a.text(x=.95, y=.95, s=f"$\Delta KL$={llik_diff.loc[k]:.1f}", horizontalalignment='right', verticalalignment='top', transform=a.transAxes) for y in range(ax.shape[0]): ax[y][0].set_ylabel('Num cells') for x in range(ax.shape[1]): ax[-1][x].set_xlabel('Num mols') fig.tight_layout()
dropseq = data['dropseq']() llik_diff = benchmark['dropseq']['npmle'] - benchmark['dropseq']['unimodal'] query = benchmark['dropseq'].loc[llik_diff[llik_diff > 0].sort_values(ascending=False).head(n=12).index].index plt.clf() fig, ax = plt.subplots(4, 3) fig.set_size_inches(7, 5) for a, k in zip(ax.ravel(), query): a.hist(dropseq[:,k], bins=np.arange(dropseq[:,k].max() + 2), color='k') a.text(x=.95, y=.95, s=f"$\Delta KL$={llik_diff.loc[k]:.1f}", horizontalalignment='right', verticalalignment='top', transform=a.transAxes) for y in range(ax.shape[0]): ax[y][0].set_ylabel('Num cells') for x in range(ax.shape[1]): ax[-1][x].set_xlabel('Num mols') fig.tight_layout()
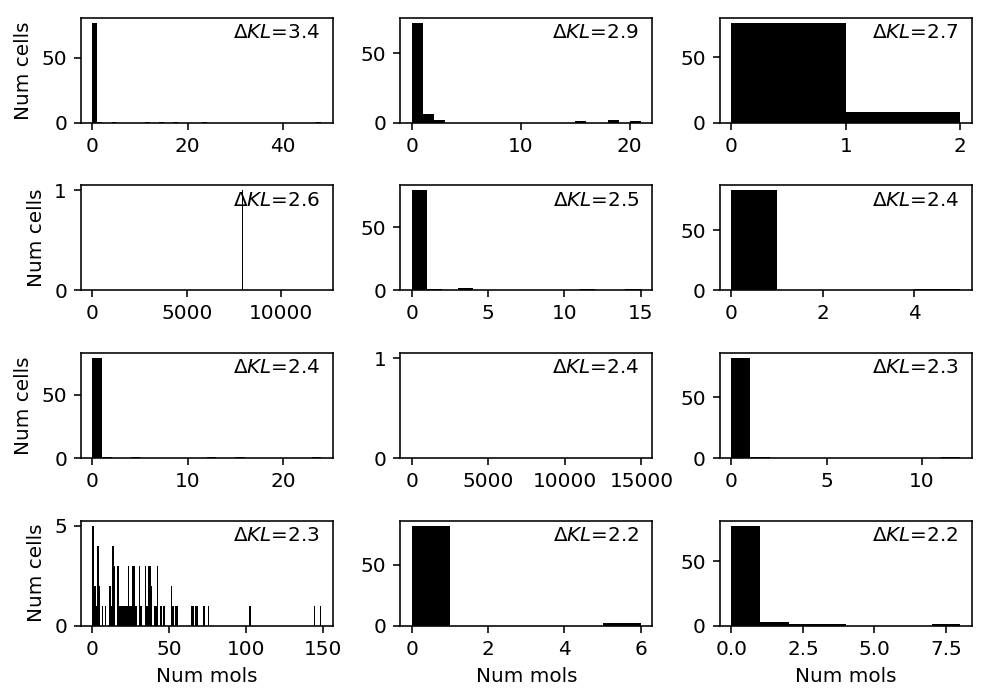
gemcode = data['gemcode']() llik_diff = benchmark['gemcode']['npmle'] - benchmark['gemcode']['unimodal'] query = llik_diff[np.logical_and(gemcode.sum(axis=0) > 0, llik_diff > 0)].sort_values(ascending=False).head(n=12).index plt.clf() fig, ax = plt.subplots(4, 3) fig.set_size_inches(7, 5) for a, k in zip(ax.ravel(), query): a.hist(gemcode[:,k], bins=np.arange(gemcode[:,k].max() + 2), color='k') a.text(x=.95, y=.95, s=f"$\Delta KL$={llik_diff.loc[k]:.1f}", horizontalalignment='right', verticalalignment='top', transform=a.transAxes) for y in range(ax.shape[0]): ax[y][0].set_ylabel('Num cells') for x in range(ax.shape[1]): ax[-1][x].set_xlabel('Num mols') fig.tight_layout()
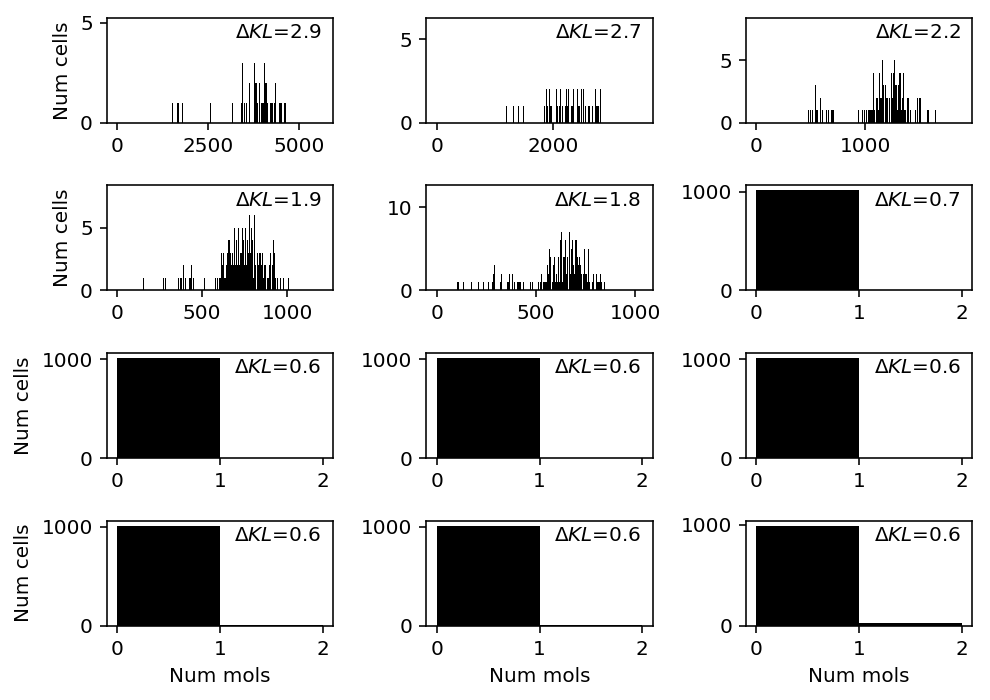
indrops = data['indrops']() llik_diff = benchmark['indrops']['npmle'] - benchmark['indrops']['unimodal'] query = llik_diff[np.logical_and(indrops.sum(axis=0) > 0, llik_diff > 0)].sort_values(ascending=False).head(n=12).index plt.clf() fig, ax = plt.subplots(4, 3) fig.set_size_inches(7, 5) for a, k in zip(ax.ravel(), query): a.hist(indrops[:,k], bins=np.arange(indrops[:,k].max() + 2), color='k') a.text(x=.95, y=.95, s=f"$\Delta KL$={llik_diff.loc[k]:.1f}", horizontalalignment='right', verticalalignment='top', transform=a.transAxes) for y in range(ax.shape[0]): ax[y][0].set_ylabel('Num cells') for x in range(ax.shape[1]): ax[-1][x].set_xlabel('Num mols') fig.tight_layout()
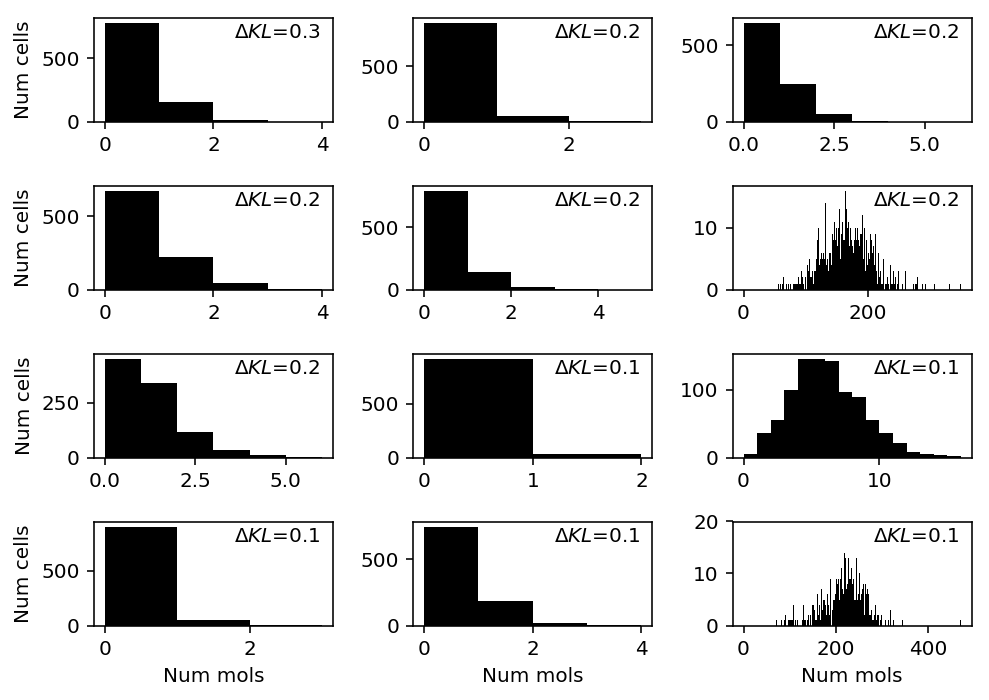
Homogeneous populations
Read the results.
benchmark = { k: g.reset_index(level=0, drop=True) for k, g in (pd.read_csv('/project2/mstephens/aksarkar/projects/singlecell-modes/data/deconv-generalization/homogeneous-kl.txt.gz', sep='\t', index_col=[0, 1]) .groupby(level=0))}
Focus on cases where NPMLE does better than unimodal. Look at B cells.
b_cells = scmodes.dataset.read_10x('/project2/mstephens/aksarkar/projects/singlecell-ideas/data/10xgenomics/b_cells/filtered_matrices_mex/hg19/', return_df=True) llik_diff = benchmark['b_cells']['npmle'] - benchmark['b_cells']['unimodal'] query = llik_diff[llik_diff > 0].sort_values(ascending=False).head(n=12).index
plt.clf() fig, ax = plt.subplots(3, 4) fig.set_size_inches(7, 5) for a, k in zip(ax.ravel(), query): a.hist(b_cells.loc[:,k], bins=np.arange(b_cells.loc[:,k].max() + 1), color='k') a.text(x=.95, y=.95, s=f"$\Delta KL$={llik_diff.loc[k]:.1g}", horizontalalignment='right', verticalalignment='top', transform=a.transAxes) a.set_title(gene_info.loc[k, 'name'] if k in gene_info.index else k) for y in range(ax.shape[0]): ax[y][0].set_ylabel('Num cells') for x in range(ax.shape[1]): ax[-1][x].set_xlabel('Num mols') fig.tight_layout()
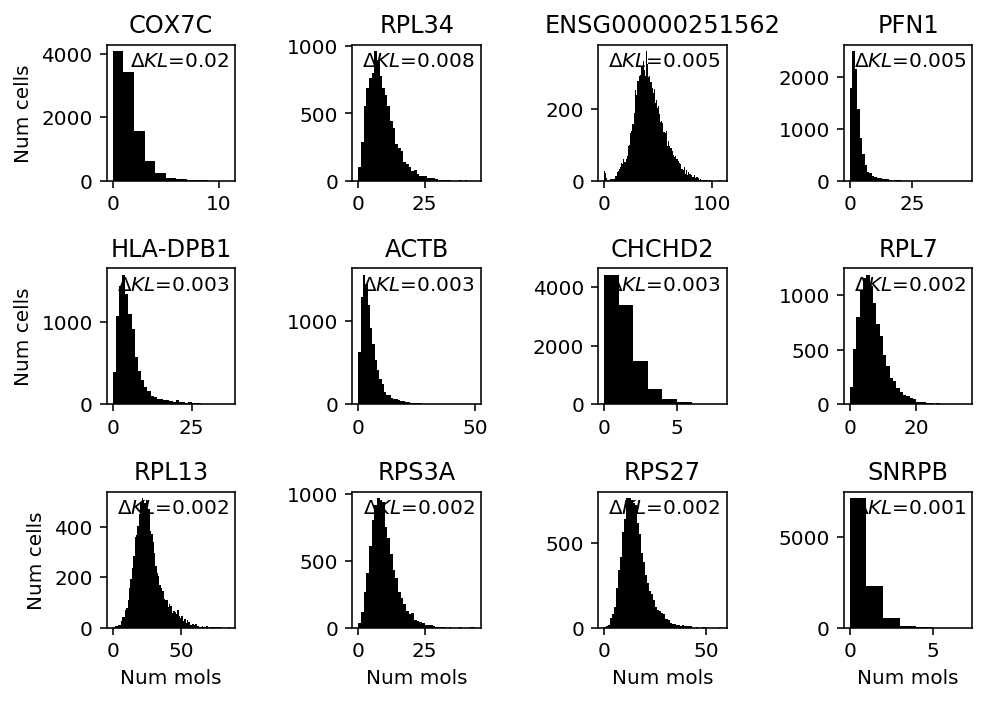
Look at cytotoxic T cells.
cytotoxic_t = scmodes.dataset.read_10x('/project2/mstephens/aksarkar/projects/singlecell-ideas/data/10xgenomics/cytotoxic_t/filtered_matrices_mex/hg19/', return_df=True) llik_diff = benchmark['cytotoxic_t']['npmle'] - benchmark['cytotoxic_t']['unimodal'] query = llik_diff[llik_diff > 0].sort_values(ascending=False).head(n=12).index
plt.clf() fig, ax = plt.subplots(4, 3) fig.set_size_inches(7, 5) for a, k in zip(ax.ravel(), query): a.hist(cytotoxic_t.loc[:,k], bins=np.arange(cytotoxic_t.loc[:,k].max() + 1), color='k') a.text(x=.95, y=.95, s=f"$\Delta KL$={llik_diff.loc[k]:.1g}", horizontalalignment='right', verticalalignment='top', transform=a.transAxes) a.set_title(gene_info.loc[k, 'name'] if k in gene_info.index else k) for y in range(ax.shape[0]): ax[y][0].set_ylabel('Num cells') for x in range(ax.shape[1]): ax[-1][x].set_xlabel('Num mols') fig.tight_layout()
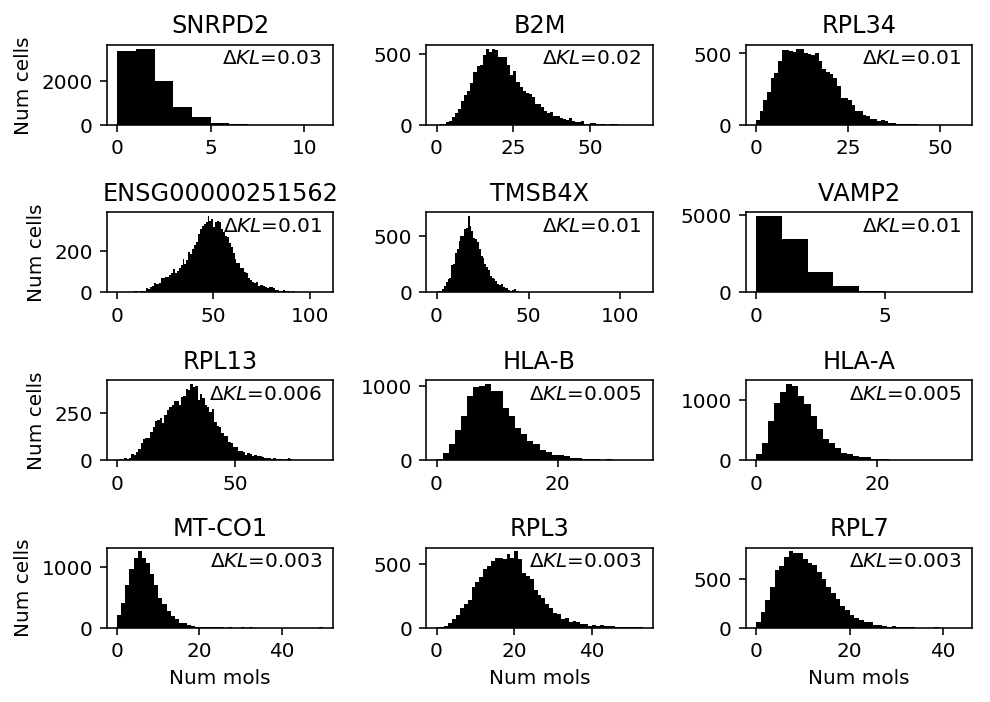
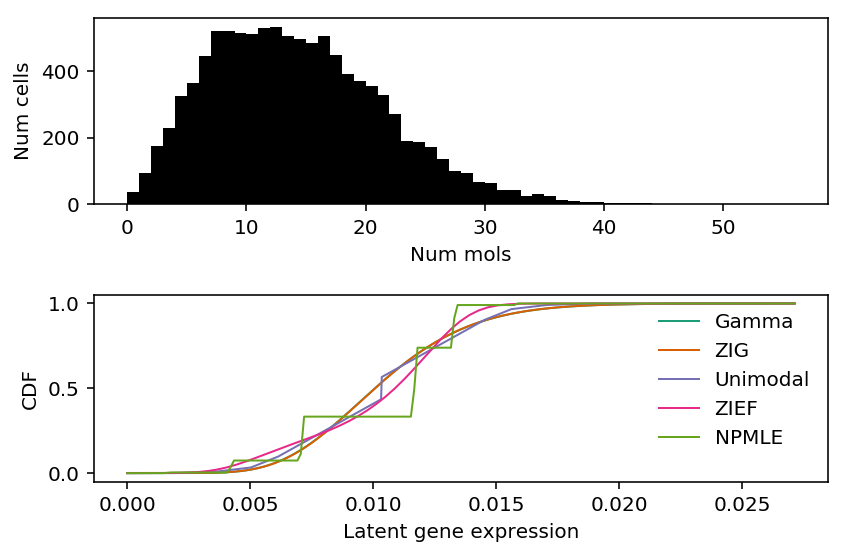
Make sure our choice of discretization for unimodal is acceptable on RPL34.
k = 'ENSG00000109475' x = cytotoxic_t.loc[:,k] s = cytotoxic_t.sum(axis=1) plt.clf() fig, ax = plt.subplots(2, 1) fig.set_size_inches(6, 4) ax[0].hist(x, bins=np.arange(x.max() + 1), color='k') ax[0].set_xlabel('Num mols') ax[0].set_ylabel('Num cells') for c, k in zip(plt.get_cmap('Dark2').colors, ['Gamma', 'ZIG', 'Unimodal', 'ZIEF', 'NPMLE']): ax[1].plot(*getattr(scmodes.deconvolve, f'fit_{k.lower()}')(x, s), color=c, lw=1, label=k) ax[1].set_xlabel('Latent gene expression') ax[1].set_ylabel('CDF') ax[1].legend(frameon=False) fig.tight_layout()
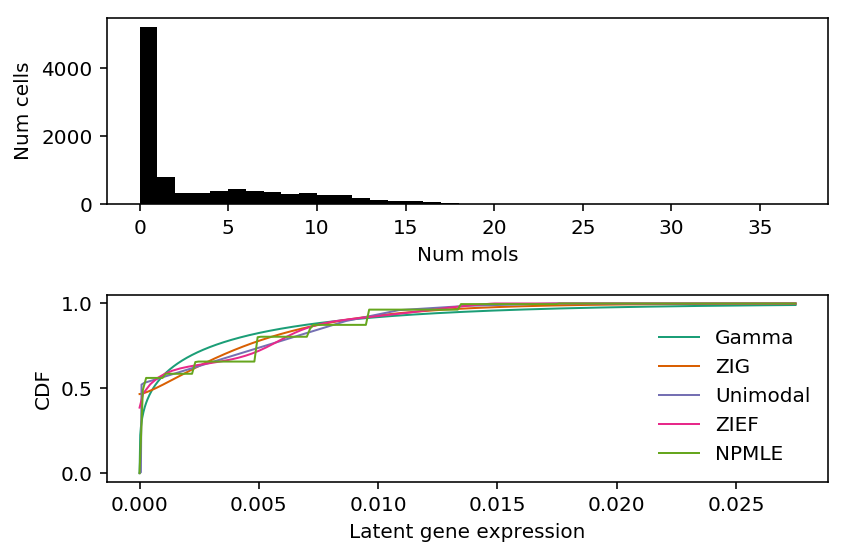
Mengyin Lu previously reported CCL5 could possibly have bimodal expression. Look at the fitted distributions for CCL5.
k = 'ENSG00000161570' x = cytotoxic_t.loc[:,k] s = cytotoxic_t.sum(axis=1) plt.clf() fig, ax = plt.subplots(2, 1) fig.set_size_inches(6, 4) ax[0].hist(x, bins=np.arange(x.max() + 1), color='k') ax[0].set_xlabel('Num mols') ax[0].set_ylabel('Num cells') for c, k in zip(plt.get_cmap('Dark2').colors, ['Gamma', 'ZIG', 'Unimodal', 'ZIEF', 'NPMLE']): ax[1].plot(*getattr(scmodes.deconvolve, f'fit_{k.lower()}')(x, s), color=c, lw=1, label=k) ax[1].set_xlabel('Latent gene expression') ax[1].set_ylabel('CDF') ax[1].legend(frameon=False) fig.tight_layout()
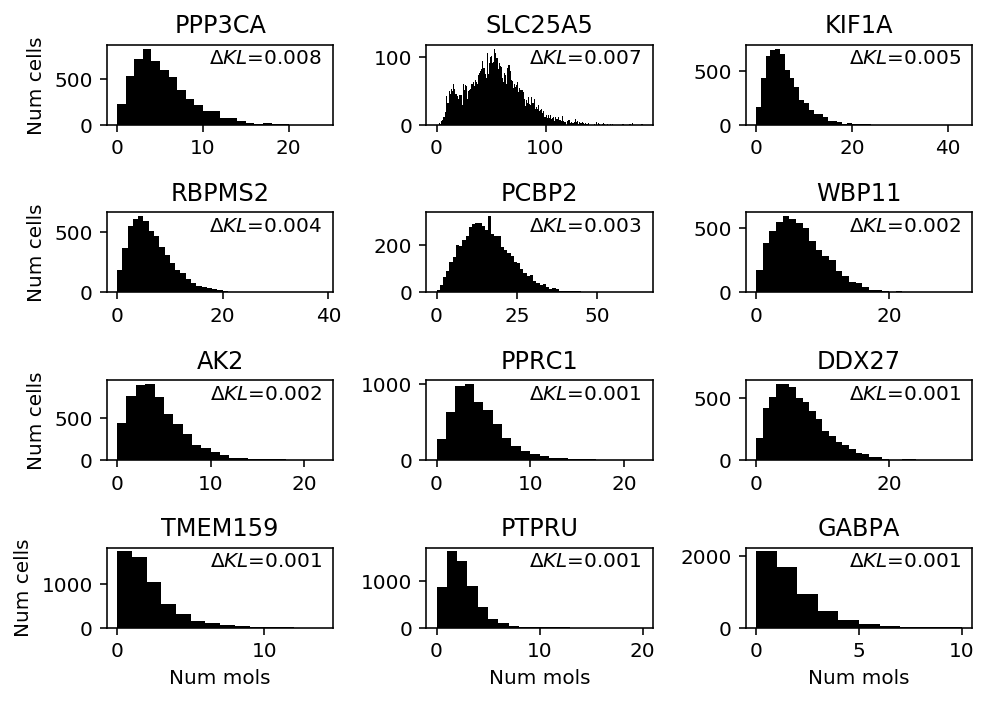
Look at iPSCs.
ipsc_counts = scmodes.dataset.ipsc('/project2/mstephens/aksarkar/projects/singlecell-qtl/data/', n=100, return_df=True) llik_diff = benchmark['ipsc']['npmle'] - benchmark['ipsc']['unimodal'] query = llik_diff[llik_diff > 0].sort_values(ascending=False).head(n=12).index
plt.clf() fig, ax = plt.subplots(4, 3) fig.set_size_inches(7, 5) for a, k in zip(ax.ravel(), query): h = a.hist(ipsc_counts.loc[:,k], bins=np.arange(ipsc_counts.loc[:,k].max() + 1), color='k') a.text(x=.95, y=.95, s=f"$\Delta KL$={llik_diff.loc[k]:.1g}", horizontalalignment='right', verticalalignment='top', transform=a.transAxes) a.set_title(gene_info.loc[k, 'name']) for y in range(ax.shape[0]): ax[y][0].set_ylabel('Num cells') for x in range(ax.shape[1]): ax[-1][x].set_xlabel('Num mols') fig.tight_layout()
Compare against Gamma.
llik_diff = benchmark['ipsc']['npmle'] - benchmark['ipsc']['gamma'] query = llik_diff[llik_diff > 0].sort_values(ascending=False).head(n=12).index plt.clf() fig, ax = plt.subplots(4, 3) fig.set_size_inches(7, 5) for a, k in zip(ax.ravel(), query): a.hist(ipsc_counts.loc[:,k], bins=np.arange(ipsc_counts.loc[:,k].max() + 1), color='k') a.text(x=.95, y=.95, s=f"$\Delta KL$={llik_diff.loc[k]:.1g}", horizontalalignment='right', verticalalignment='top', transform=a.transAxes) a.set_title(gene_info.loc[k, 'name']) for y in range(ax.shape[0]): ax[y][0].set_ylabel('Num cells') for x in range(ax.shape[1]): ax[-1][x].set_xlabel('Num mols') fig.tight_layout()
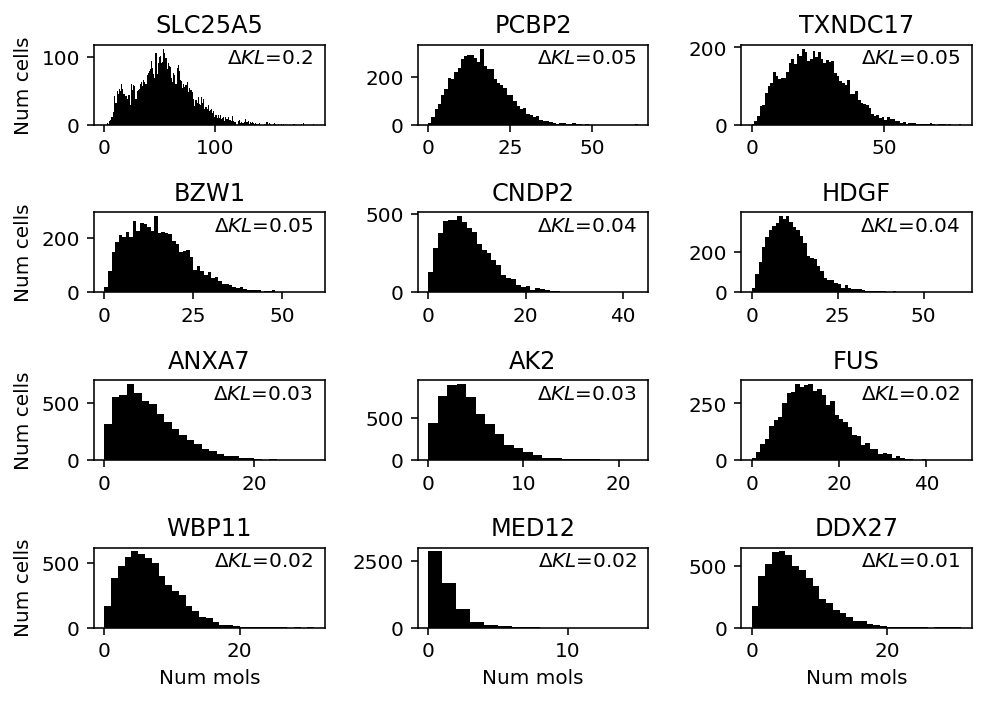
- SLC25A5 (Solute carrier family 25 member 5) codes for a mitochrondrial membrane protein (Rocha et al. 2018), and is altered in Parkinson's disease
- TXNDC17 (Thioredoxin domain-containing protein 17) appears to be involved in oxidative stress
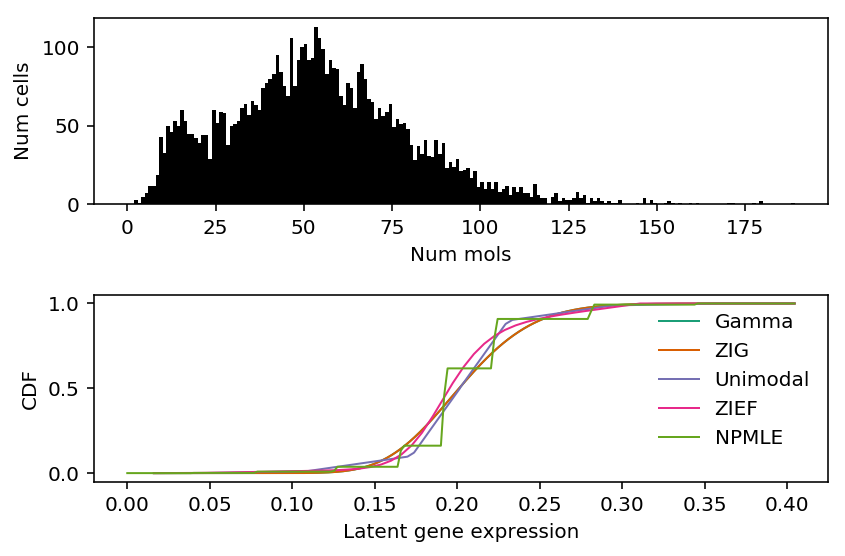
Look at the fitted distributions for SLC25A5.
k = 'ENSG00000005022' x = ipsc_counts.loc[:,k] s = ipsc_counts.sum(axis=1) plt.clf() fig, ax = plt.subplots(2, 1) fig.set_size_inches(6, 4) ax[0].hist(x, bins=np.arange(x.max() + 1), color='k') ax[0].set_xlabel('Num mols') ax[0].set_ylabel('Num cells') for c, k in zip(plt.get_cmap('Dark2').colors, ['Gamma', 'ZIG', 'Unimodal', 'ZIEF', 'NPMLE']): ax[1].plot(*getattr(scmodes.deconvolve, f'fit_{k.lower()}')(x, s), color=c, lw=1, label=k) ax[1].set_xlabel('Latent gene expression') ax[1].set_ylabel('CDF') ax[1].legend(frameon=False) fig.tight_layout()
Conclusions:
- When NPMLE does better than Gamma, unimodal does comparably well. The genes where this happens quite clearly exhibit unimodal gene expression.
Investigate Gamma vs. Point-Gamma in high-depth data
Look at the full benchmark results for NB vs. ZINB (only the GPU implementation is suitable for this).
ipsc_res = pd.read_csv('/project2/mstephens/aksarkar/projects/singlecell-modes/data/deconv-generalization/ipsc-gpu.txt.gz', index_col=0, sep='\t')
plt.clf() plt.gcf().set_size_inches(3, 3) h = plt.hist(ipsc_res['zinb'] - ipsc_res['nb'], color='k', bins=np.arange(-10, 40, 2)) plt.xlabel('Diff val set log lik from NB') _ = plt.ylabel('Number of genes')
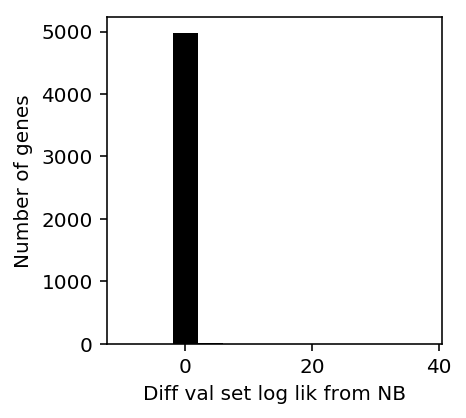
Print the histogram for inspection.
np.vstack((h[1][:-1], h[0])).T.astype(int)
array([[ -10, 0], [ -8, 0], [ -6, 0], [ -4, 0], [ -2, 4977], [ 0, 4973], [ 2, 2], [ 4, 3], [ 6, 0], [ 8, 0], [ 10, 0], [ 12, 0], [ 14, 1], [ 16, 0], [ 18, 0], [ 20, 0], [ 22, 0], [ 24, 0], [ 26, 0], [ 28, 1], [ 30, 0], [ 32, 0], [ 34, 0], [ 36, 0]])
Try to find a case in the full iPSC data where point-Gamma (ZINB) is required, by performing likelihood ratio tests against Gamma (NB). Use Bonferroni correction to control FWER.
lrt = st.chi2(1).sf(-2 * (ipsc_res['gamma'] - ipsc_res['point_gamma'])) ipsc_res[lrt < .05 / lrt.shape[0]]
gamma point_gamma gene ENSG00000112530 -2017.717750 -1987.894352 ENSG00000129824 -1803.651932 -1789.106410
The two genes are PACRG and RPS4Y1. Look at the fitted distributions for these genes.
ipsc_counts = scmodes.dataset.ipsc(prefix='/project2/mstephens/aksarkar/projects/singlecell-qtl/data/', query=list(ipsc_res[lrt < .05 / lrt.shape[0]].index), return_df=True)
annotation = pd.read_csv('/project2/mstephens/aksarkar/projects/singlecell-qtl/data/scqtl-annotation.txt', sep='\t') keep_samples = pd.read_csv('/project2/mstephens/aksarkar/projects/singlecell-qtl/data/quality-single-cells.txt', index_col=0, header=None, sep='\t') s = annotation.loc[keep_samples.values.ravel(), 'mol_hs']
k = 'ENSG00000112530' x = ipsc_counts.loc[:,k].values plt.clf() fig, ax = plt.subplots(2, 1) fig.set_size_inches(6, 4) ax[0].hist(x, bins=np.arange(x.max() + 1), color='k') ax[0].set_title(gene_info.loc[k, 'name']) ax[0].set_xlabel('Num mols') ax[0].set_ylabel('Num cells') for c, k in zip(plt.get_cmap('Dark2').colors, ['Gamma', 'ZIG', 'Unimodal', 'ZIEF', 'NPMLE']): ax[1].plot(*getattr(scmodes.deconvolve, f'fit_{k.lower()}')(x, s), color=c, lw=1, label=k) ax[1].set_xlabel('Latent gene expression') ax[1].set_ylabel('CDF') ax[1].legend(frameon=False) fig.tight_layout()
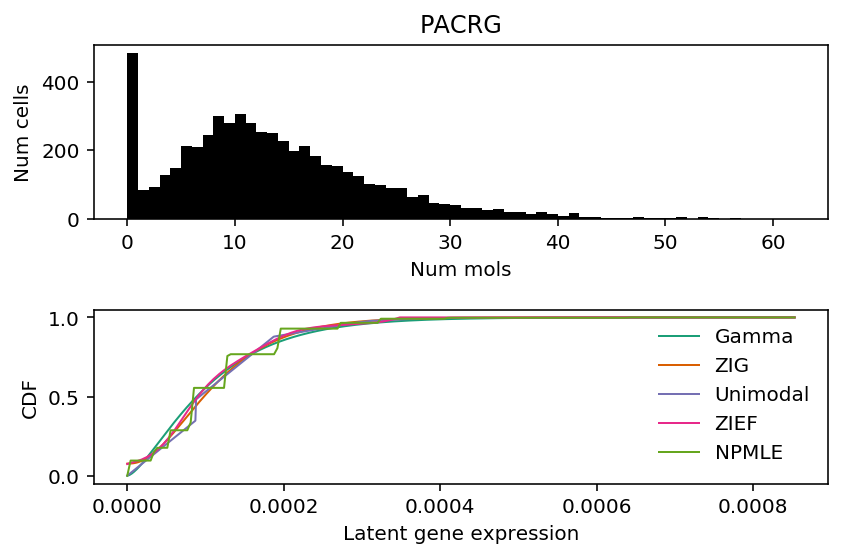
PACRG (Parkin coregulated gene) shares a promoter with PKRG (PARK2; Parkin). PARKIN is involved in a signaling pathway for mitochondria damage, and mitochrondrial damage appears to be relevant to neurodegenerative disease etiology (Alzheimer's disease and Parkinson's disease). Mitochrondrial damage could explain the bimodal distribution of PACRG in the iPSC data: the reprogramming protocol could induce oxidative stress on some of the cells, resulting in damage.
k = 'ENSG00000129824' x = ipsc_counts.loc[:,k].values plt.clf() fig, ax = plt.subplots(2, 1) fig.set_size_inches(6, 4) ax[0].hist(x, bins=np.arange(x.max() + 1), color='k') ax[0].set_title(gene_info.loc[k, 'name']) ax[0].set_xlabel('Num mols') ax[0].set_ylabel('Num cells') for c, k in zip(plt.get_cmap('Dark2').colors, ['Gamma', 'ZIG', 'Unimodal', 'ZIEF', 'NPMLE']): ax[1].plot(*getattr(scmodes.deconvolve, f'fit_{k.lower()}')(x, s), color=c, lw=1, label=k) ax[1].set_xlabel('Latent gene expression') ax[1].set_ylabel('CDF') ax[1].legend(frameon=False) fig.tight_layout()
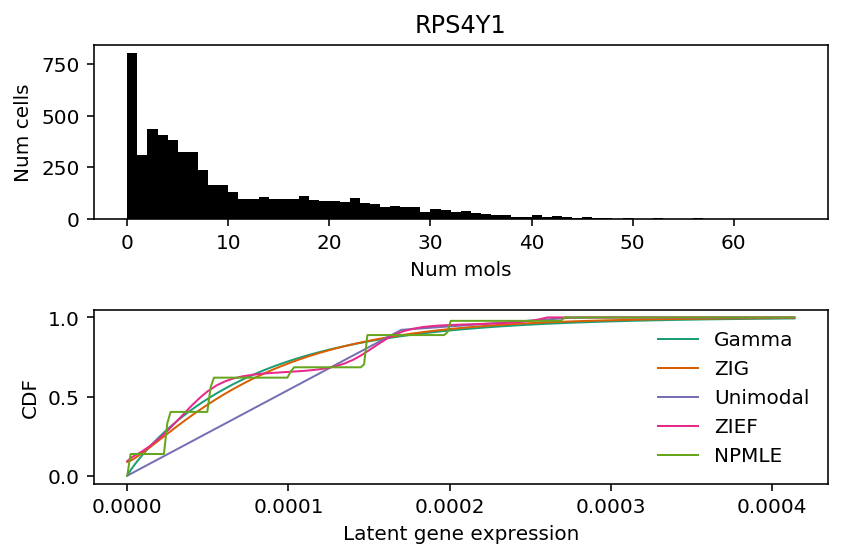
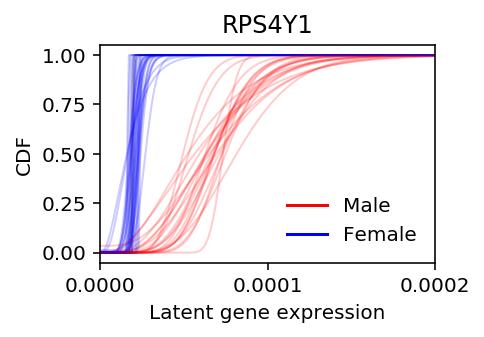
RPS4Y1 (Ribosomal protein S4, Y-linked) is on the Y chromosome, and therefore we should expect it to only be expressed in males. The homologous gene on the X chromosome (RPS4X) has a different coding sequence. Naively, sex-linkage should explain why this gene exhibits bimodal gene expression.
curl -OL 'http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/working/20130606_sample_info/20130606_sample_info.txt'
donor_info = pd.read_csv('/project2/mstephens/aksarkar/projects/singlecell-modes/data/20130606_sample_info.txt', sep='\t')
Count how many cells come from females.
prefix = '/project2/mstephens/aksarkar/projects/singlecell-qtl/data/' annotations = pd.read_csv(f'{prefix}/scqtl-annotation.txt', sep='\t') keep_samples = pd.read_csv(f'{prefix}/quality-single-cells.txt', sep='\t', index_col=0, header=None) annotations = annotations.loc[keep_samples.values.ravel()]
annotations.merge(donor_info, left_on='chip_id', right_on='Sample')['Gender'].value_counts()
female 3109 male 2369 Name: Gender, dtype: int64
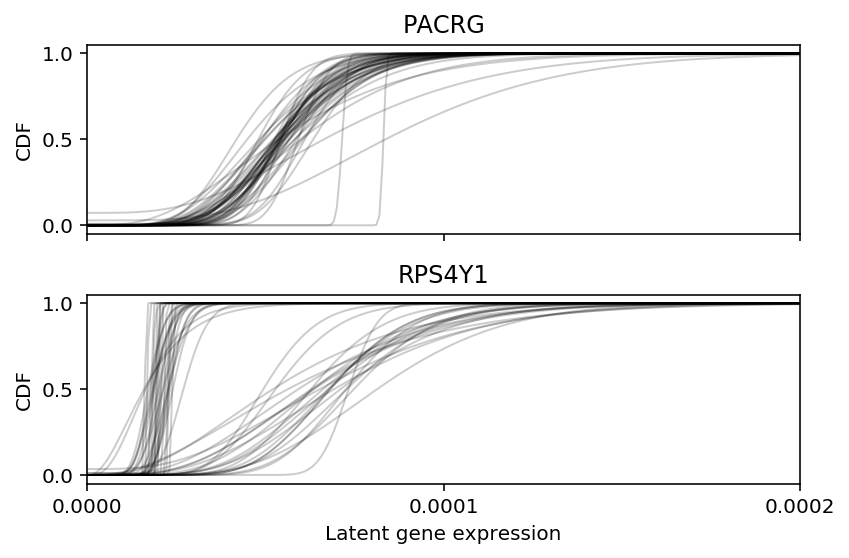
Look at the fitted ZINB distribution per-individual for this gene.
ipsc_log_mu = pd.read_csv('/project2/mstephens/aksarkar/projects/singlecell-qtl/data/density-estimation/design1/zi2-log-mu.txt.gz', sep=' ', index_col=0) ipsc_log_phi = pd.read_csv('/project2/mstephens/aksarkar/projects/singlecell-qtl/data/density-estimation/design1/zi2-log-phi.txt.gz', sep=' ', index_col=0) ipsc_logodds = pd.read_csv('/project2/mstephens/aksarkar/projects/singlecell-qtl/data/density-estimation/design1/zi2-logodds.txt.gz', sep=' ', index_col=0)
def point_gamma_cdf(grid, log_mu, log_phi, logodds): res = st.gamma(a=np.exp(-log_phi), scale=np.exp(log_mu + log_phi)).cdf(grid) res *= sp.expit(-logodds) res += sp.expit(logodds) return res lam = ipsc_counts.values / annotations['mol_hs'].values.reshape(-1, 1) grid = np.linspace(lam.min(), lam.max(), 1000) cdfs = np.array([[point_gamma_cdf(grid, ipsc_log_mu.loc[j, k], ipsc_log_phi.loc[j, k], ipsc_logodds.loc[j, k]) for k in ipsc_log_mu] for j in ipsc_counts.columns if j != list(ipsc_log_mu.columns).index('NA18498')])
plt.clf() fig, ax = plt.subplots(2, 1, sharex=True) fig.set_size_inches(6, 4) for a, F, k in zip(ax, cdfs, ipsc_counts.columns): for f in F: a.set_xlim(0, 2e-4) a.set_xticks(np.linspace(0, 2e-4, 3)) a.plot(grid, f, c='k', lw=1, alpha=0.2) a.set_title(gene_info.loc[k, 'name']) a.set_ylabel('CDF') ax[-1].set_xlabel('Latent gene expression') fig.tight_layout()
Plot the estimated RPS4Y1 latent gene expression, stratified by donor sex.
colors = {k: 'r' if v == 'male' else 'b' for k, v in donor_info.set_index('Sample') .filter(items=list(ipsc_log_mu.columns), axis=0) ['Gender'] .iteritems()} plt.clf() plt.gcf().set_size_inches(3, 2) for f, k in zip(cdfs[1], ipsc_log_mu.columns): plt.xlim(0, 2e-4) plt.xticks(np.linspace(0, 2e-4, 3)) plt.plot(grid, f, lw=1, alpha=0.2, c=colors.get(k, 'k')) dummy = [plt.Line2D([0], [0], c='r'), plt.Line2D([0], [0], c='b')] plt.legend(dummy, ['Male', 'Female'], frameon=False) plt.title('RPS4Y1') plt.ylabel('CDF') plt.xlabel('Latent gene expression')
Synthetic mixtures
Read the results.
benchmark = { k: g.reset_index(level=0, drop=True) for k, g in (pd.read_csv('/project2/mstephens/aksarkar/projects/singlecell-modes/data/deconv-generalization/synthetic-mix-kl.txt.gz', sep='\t', index_col=[0, 1]) .groupby(level=0))}
Read the data.
t_cells = scmodes.dataset.read_10x('/project2/mstephens/aksarkar/projects/singlecell-ideas/data/10xgenomics/cytotoxic_t/filtered_matrices_mex/hg19/', min_detect=0, return_df=True) b_cells = scmodes.dataset.read_10x('/project2/mstephens/aksarkar/projects/singlecell-ideas/data/10xgenomics/b_cells/filtered_matrices_mex/hg19/', min_detect=0, return_df=True) mix1, y1 = scmodes.dataset.synthetic_mix(t_cells, b_cells, min_detect=0.25) cyto = scmodes.dataset.read_10x(prefix='/project2/mstephens/aksarkar/projects/singlecell-ideas/data/10xgenomics/cytotoxic_t/filtered_matrices_mex/hg19', return_df=True, min_detect=0) naive = scmodes.dataset.read_10x(prefix='/project2/mstephens/aksarkar/projects/singlecell-ideas/data/10xgenomics/naive_cytotoxic/filtered_matrices_mex/hg19', return_df=True, min_detect=0) mix2, y2 = scmodes.dataset.synthetic_mix(cyto, naive, min_detect=0.25)
Look at T cell/B cell mix.
llik_diff = benchmark['cd8-cd19-mix']['npmle'] - benchmark['cd8-cd19-mix']['unimodal'] query = benchmark['cd8-cd19-mix'].loc[llik_diff[llik_diff > 0].sort_values(ascending=False).head(n=12).index].index.astype(int) plt.clf() fig, ax = plt.subplots(3, 4) fig.set_size_inches(8, 5) for a, k in zip(ax.ravel(), query): h = a.hist(mix1.loc[y1 == 1,mix1.columns[k]], bins=np.arange(mix1.iloc[:,k].max() + 1), color='k', alpha=0.6, label='T cells') a.hist(mix1.loc[y1 == 0,mix1.columns[k]], bins=np.arange(mix1.iloc[:,k].max() + 1), color='r', alpha=0.6, label='B cells') a.text(x=.95, y=.95, s=f"$\Delta KL$={llik_diff.iloc[k]:.1g}", horizontalalignment='right', verticalalignment='top', transform=a.transAxes) a.set_title(gene_info.loc[mix1.columns[k], 'name'] if mix1.columns[k] in gene_info.index else mix1.columns[k]) handles, labels = a.get_legend_handles_labels() fig.legend(handles, labels, frameon=False, loc='center left', bbox_to_anchor=(1, 0.5)) for a in ax: a[0].set_ylabel('Num cells') for a in ax.T: a[-1].set_xlabel('Num mols') fig.tight_layout()
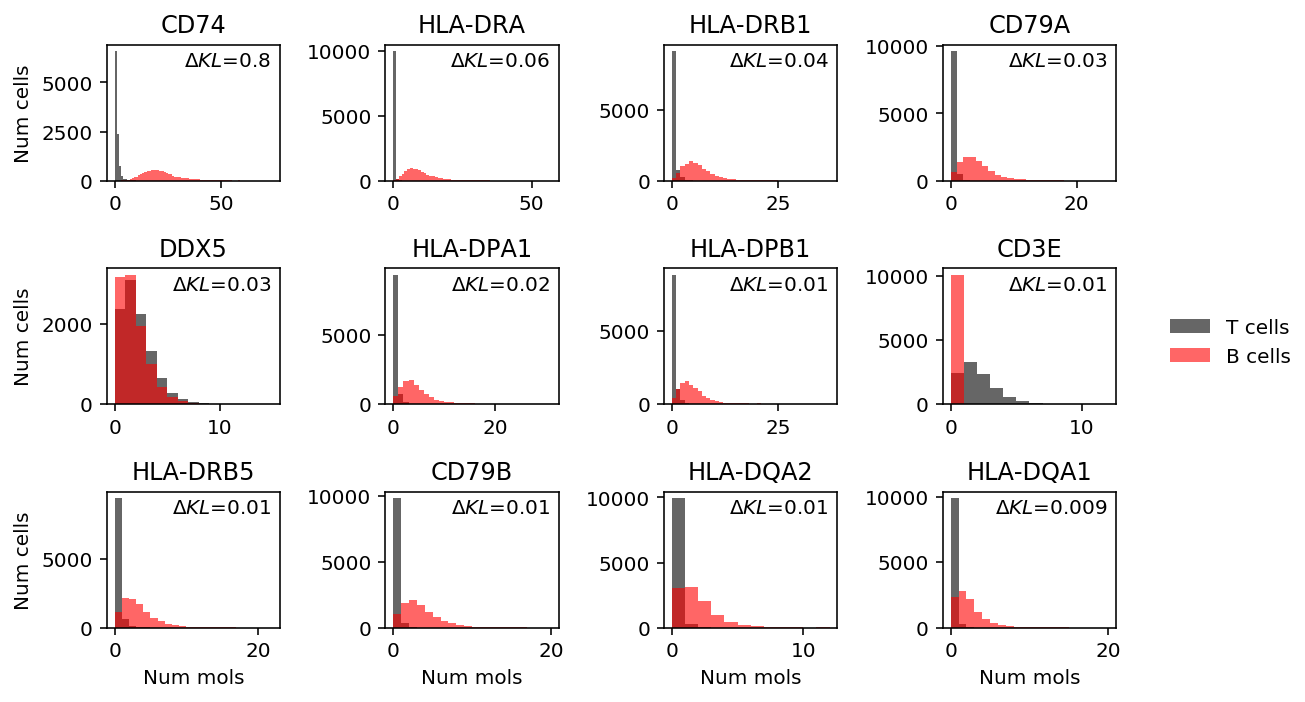
llik_diff = benchmark['cd8-cd19-mix']['npmle'] - benchmark['cd8-cd19-mix']['unimodal'] query = benchmark['cd8-cd19-mix'].loc[llik_diff[llik_diff > 0].sort_values(ascending=False).head(n=4).index].index.astype(int) plt.clf() fig, ax = plt.subplots(2, 2) fig.set_size_inches(4, 4) for a, k in zip(ax.ravel(), query): h = a.hist(mix1.loc[y1 == 1,mix1.columns[k]], bins=np.arange(mix1.iloc[:,k].max() + 1), color='k', alpha=0.6, label='T cells') a.hist(mix1.loc[y1 == 0,mix1.columns[k]], bins=np.arange(mix1.iloc[:,k].max() + 1), color='r', alpha=0.6, label='B cells') a.text(x=.95, y=.95, s=f"$\Delta l$={llik_diff.iloc[k]:.1g}", horizontalalignment='right', verticalalignment='top', transform=a.transAxes) a.set_title(gene_info.loc[mix1.columns[k], 'name'] if mix1.columns[k] in gene_info.index else mix1.columns[k]) ax[0, 0].legend(frameon=False, loc='center right') for a in ax: a[0].set_ylabel('Num cells') for a in ax.T: a[-1].set_xlabel('Num molecules') fig.tight_layout()
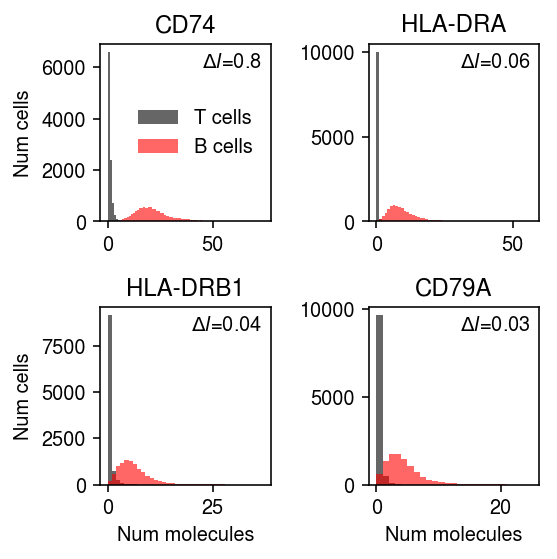
Look cytotoxic/naive T cell mix.
llik_diff = benchmark['cyto-naive-t-mix']['npmle'] - benchmark['cyto-naive-t-mix']['unimodal'] query = benchmark['cyto-naive-t-mix'].loc[llik_diff[llik_diff > 0].sort_values(ascending=False).head(n=12).index].index plt.clf() fig, ax = plt.subplots(3, 4) fig.set_size_inches(8, 5) for a, k in zip(ax.ravel(), query): a.hist(mix2.loc[y1 == 1,k], bins=np.arange(mix2.loc[:,k].max() + 1), alpha=0.6, color='k', label='Cytotoxic T') a.hist(mix2.loc[y1 == 0,k], bins=np.arange(mix2.loc[:,k].max() + 1), alpha=0.6, color='r', label='Naive T') a.text(x=.95, y=.95, s=f"$\Delta KL$={llik_diff.loc[k]:.1g}", horizontalalignment='right', verticalalignment='top', transform=a.transAxes) a.set_title(gene_info.loc[k, 'name'] if k in gene_info.index else k) handles, labels = a.get_legend_handles_labels() fig.legend(handles, labels, frameon=False, loc='center left', bbox_to_anchor=(1, 0.5)) for a in ax: a[0].set_ylabel('Num cells') for a in ax.T: a[-1].set_xlabel('Num mols') fig.tight_layout()
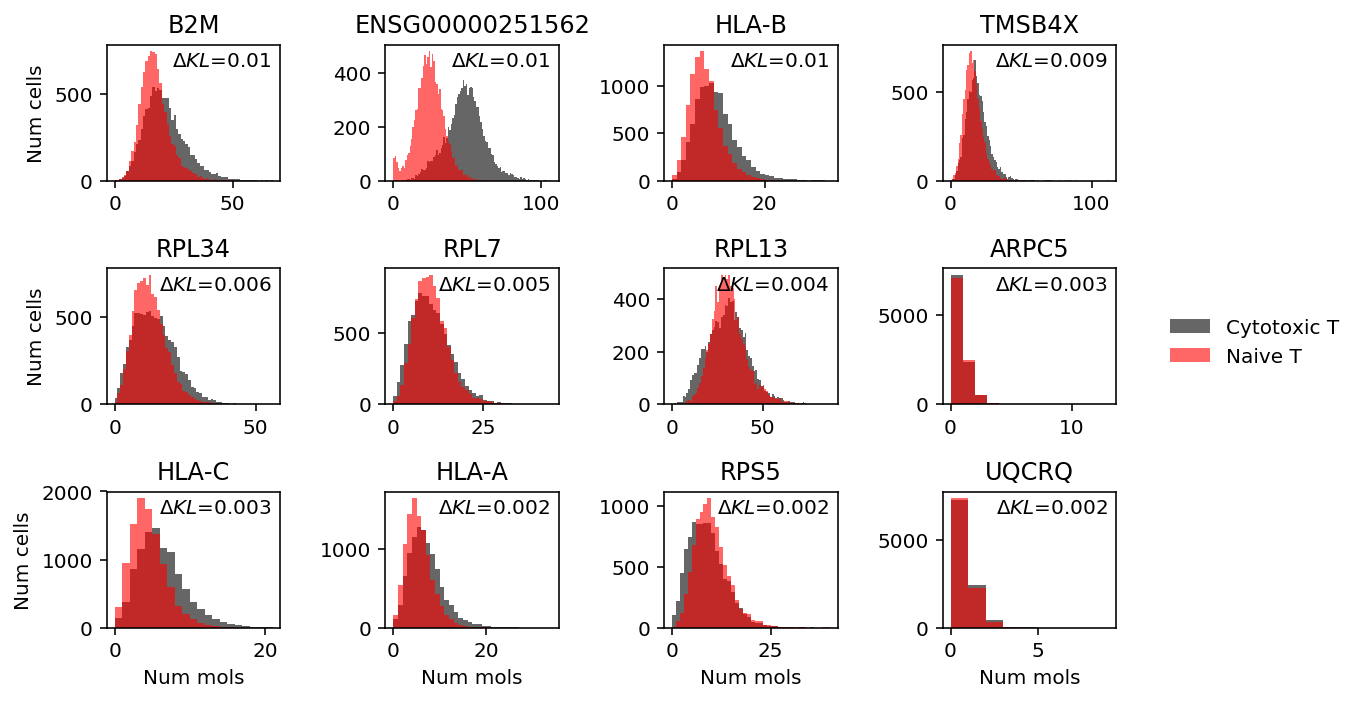
MALAT1 (Metastasis-associated lung adenocarcinoma transcript 1) is a lincRNA which appears to show tri-modal gene expression in this mixture. It is involved in T cell activation, through interaction with NF-κB (Zhao et al. 2016).
Heterogeneous populations
Read the results.
benchmark = { k: g.reset_index(level=0, drop=True) for k, g in (pd.read_csv('/project2/mstephens/aksarkar/projects/singlecell-modes/data/deconv-generalization/heterogeneous-kl.txt.gz', sep='\t', index_col=[0, 1]) .groupby(level=0))}
Cortex
Fix the cortex count matrix (seems to break the parser in pandas).
import scmodes work = scmodes.dataset.cortex('/project2/mstephens/aksarkar/projects/singlecell-ideas/data/zeisel-2015/GSE60361_C1-3005-Expression.txt.gz', return_df=True) work.to_csv('/project2/mstephens/aksarkar/projects/singlecell-ideas/data/zeisel-2015/zeisel-2015.txt.gz', compression='gzip', sep='\t')
sbatch --partition=broadwl --mem=16G #!/bin/bash source activate scmodes python <<EOF <<fix-cortex>> EOF
cortex_counts = pd.read_csv('/project2/mstephens/aksarkar/projects/singlecell-ideas/data/zeisel-2015/zeisel-2015.txt.gz', sep='\t', index_col=0)
Look at the fitted distribution for the genes where NPMLE does worst.
llik_diff = benchmark['cortex']['npmle'] - benchmark['cortex']['gamma'] llik_diff.sort_values().head(n=12)
Ttr -3.136362 Hbb-bs -3.032071 Hba-a2_loc1 -2.053220 Hba-a2_loc2 -1.985382 Sst -1.842792 Apoe -1.414460 Npy -1.403264 Hbb-b2 -1.365434 Ccl3 -1.226098 Mgp -1.198429 Acta2 -1.139285 Ptgds -1.132885 dtype: float64
k = 'Ttr' x = cortex_counts.loc[:,k].values s = cortex_counts.sum(axis=1).values plt.clf() fig, ax = plt.subplots(2, 1) fig.set_size_inches(6, 4) ax[0].hist(x, bins=np.arange(0, x.max() + 1, 100), color='k') ax[0].set_title(k.upper()) ax[0].set_xlabel('Num mols') ax[0].set_ylabel('Num cells') for c, k in zip(plt.get_cmap('Dark2').colors, ['Gamma', 'ZIG', 'Unimodal', 'ZIEF', 'NPMLE']): ax[1].plot(*getattr(scmodes.deconvolve, f'fit_{k.lower()}')(x, s), color=c, lw=1, label=k) ax[1].set_xlabel('Latent gene expression') ax[1].set_ylabel('CDF') ax[1].legend(frameon=False) fig.tight_layout()
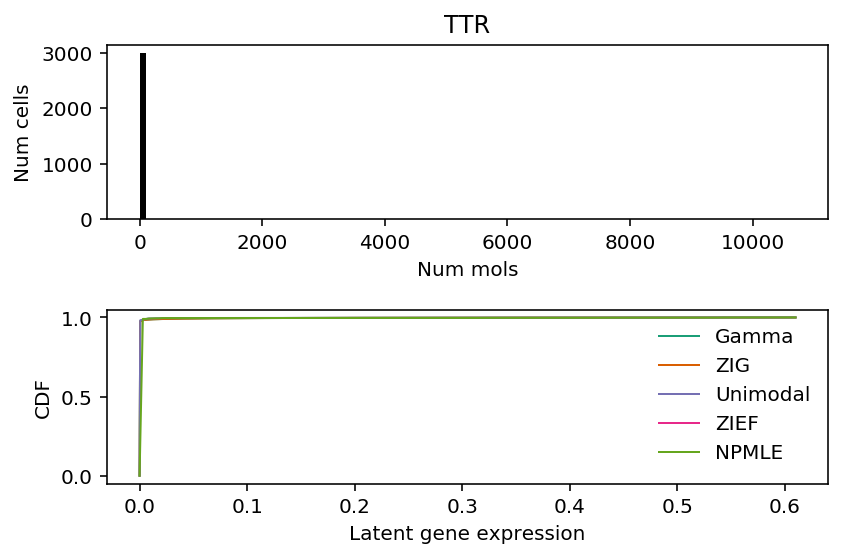
Look at the distribution of latent gene expression values (naively estimated) for TTR.
pd.Series((x / s)[x > 0]).describe()
count 90.000000 mean 0.059269 std 0.152847 min 0.000055 25% 0.000368 50% 0.003031 75% 0.008099 max 0.610003 dtype: float64
For these genes, more than 90% of the samples have observed count 0, so the optimal model appears to predict 0 for all samples.
Look at genes where ZIG does better than Gamma.
llik_diff = benchmark['cortex']['point_gamma'] - benchmark['cortex']['gamma'] query = benchmark['cortex'].loc[llik_diff[llik_diff > 0].sort_values(ascending=False).head(n=12).index].index plt.clf() fig, ax = plt.subplots(4, 3) fig.set_size_inches(7, 5) for a, k in zip(ax.ravel(), query): a.hist(cortex_counts.loc[:,k], bins=np.arange(cortex_counts.loc[:,k].max() + 1), color='k') a.text(x=.95, y=.95, s=f"$\Delta KL$={llik_diff.loc[k]:.1f}", horizontalalignment='right', verticalalignment='top', transform=a.transAxes) a.set_title(k.upper()) for y in range(ax.shape[0]): ax[y][0].set_ylabel('Num cells') for x in range(ax.shape[1]): ax[-1][x].set_xlabel('Num mols') fig.tight_layout()
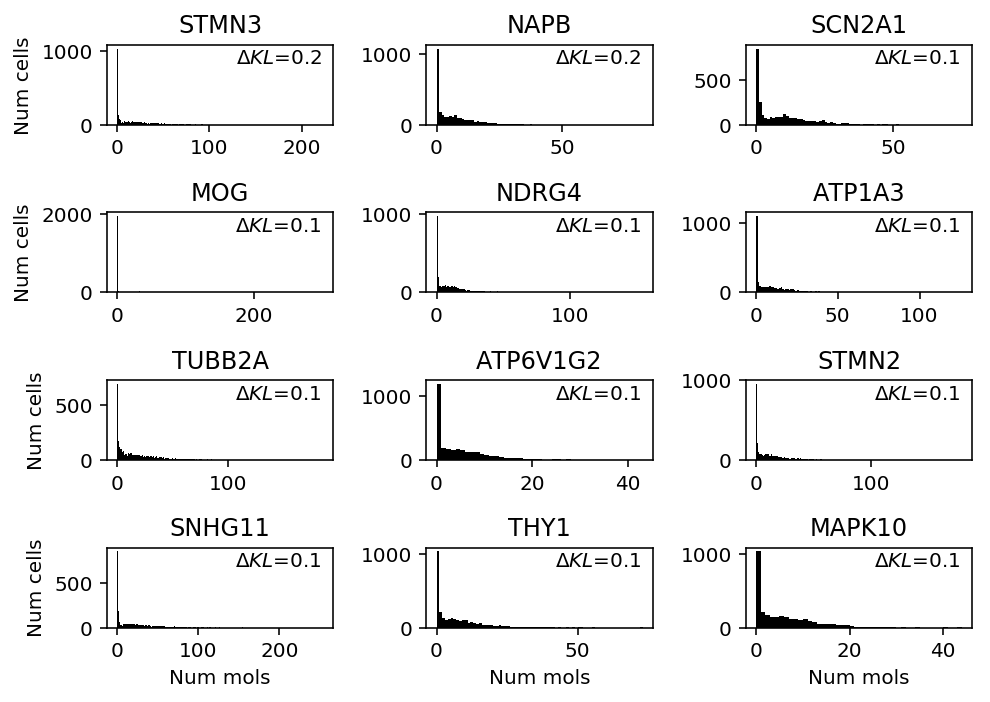
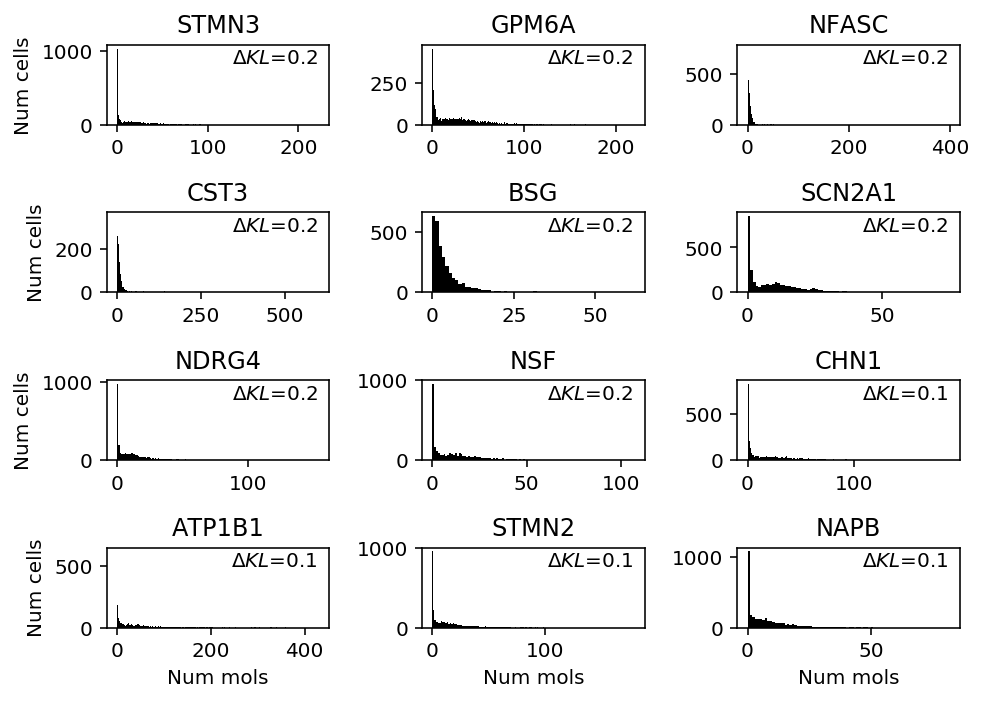
Look at genes where unimodal shows greatest performance gain.
llik_diff = benchmark['cortex']['unimodal'] - benchmark['cortex']['gamma'] query = benchmark['cortex'].loc[llik_diff[llik_diff > 0].sort_values(ascending=False).head(n=12).index].index plt.clf() fig, ax = plt.subplots(4, 3) fig.set_size_inches(7, 5) for a, k in zip(ax.ravel(), query): a.hist(cortex_counts.loc[:,k], bins=np.arange(cortex_counts.loc[:,k].max() + 1), color='k') a.text(x=.95, y=.95, s=f"$\Delta KL$={llik_diff.loc[k]:.1f}", horizontalalignment='right', verticalalignment='top', transform=a.transAxes) a.set_title(k.upper()) for y in range(ax.shape[0]): ax[y][0].set_ylabel('Num cells') for x in range(ax.shape[1]): ax[-1][x].set_xlabel('Num mols') fig.tight_layout()
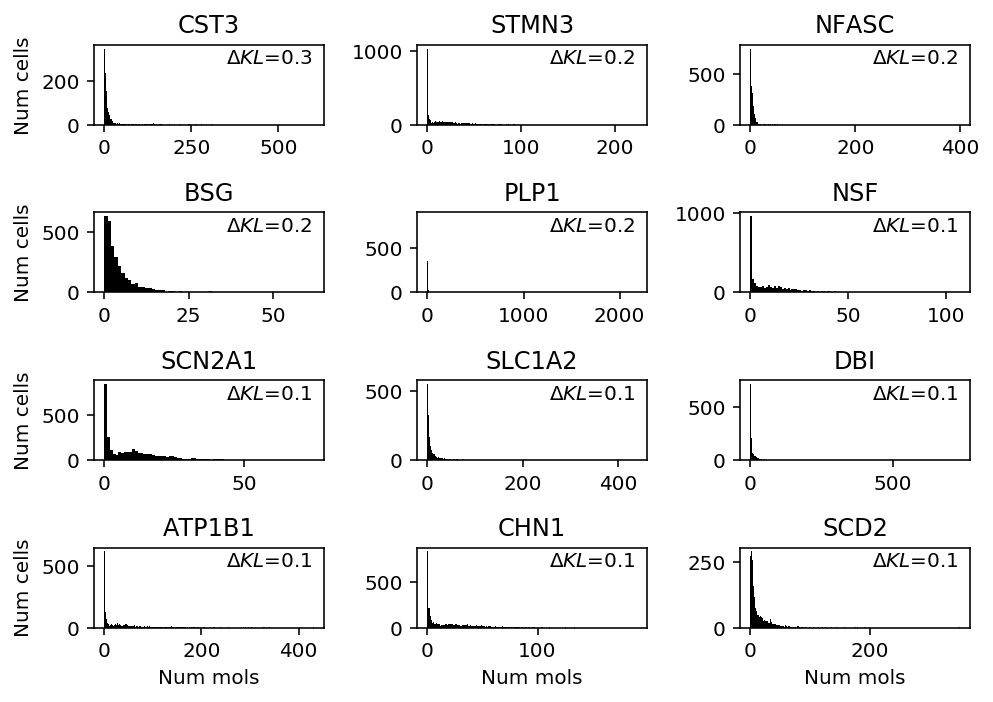
Show genes where NPMLE shows greatest performance gain.
llik_diff = benchmark['cortex']['npmle'] - benchmark['cortex']['gamma'] query = benchmark['cortex'].loc[llik_diff[llik_diff > 0].sort_values(ascending=False).head(n=12).index].index plt.clf() fig, ax = plt.subplots(4, 3) fig.set_size_inches(7, 5) for a, k in zip(ax.ravel(), query): a.hist(cortex_counts.loc[:,k], bins=np.arange(cortex_counts.loc[:,k].max() + 1), color='k') a.text(x=.95, y=.95, s=f"$\Delta KL$={llik_diff.loc[k]:.1f}", horizontalalignment='right', verticalalignment='top', transform=a.transAxes) a.set_title(k.upper()) for y in range(ax.shape[0]): ax[y][0].set_ylabel('Num cells') for x in range(ax.shape[1]): ax[-1][x].set_xlabel('Num mols') fig.tight_layout()
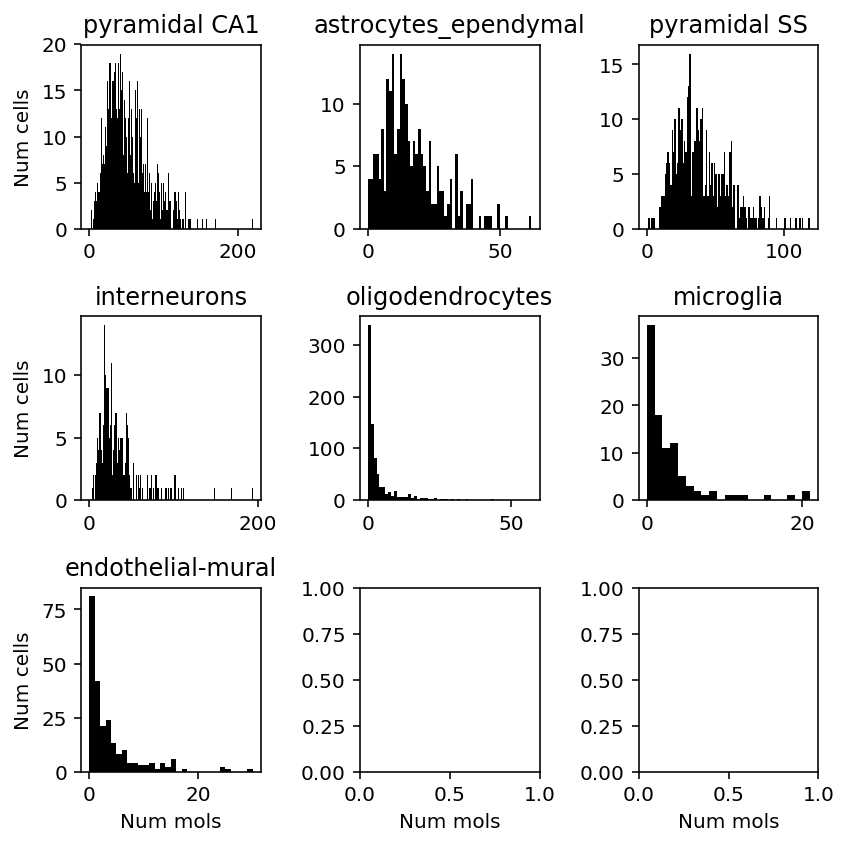
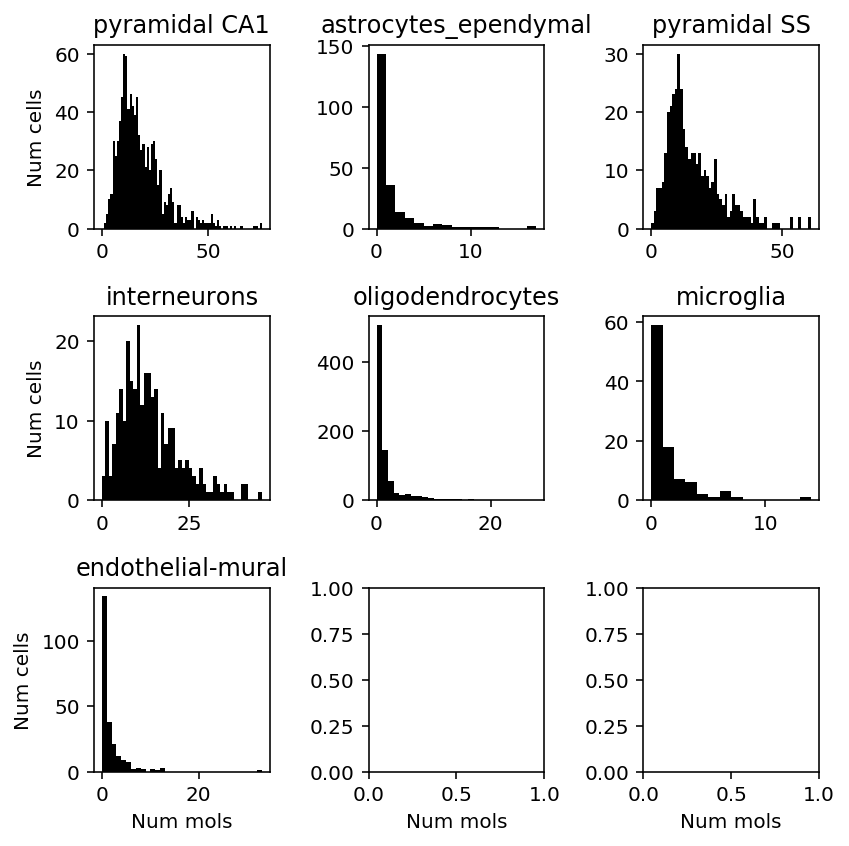
- GPM6A (Glycoprotein m6a) appears to be required for mouse brain develoment, and is expressed in specific brain regions
- SCN2A (Sodium channel, voltage-gated, type II, alpha) is needed for propagating actions potentials in neurons
Stratify the gene expression of these genes by inferred cell type.
curl -sLO "https://storage.googleapis.com/linnarsson-lab-www-blobs/blobs/cortex/expression_mRNA_17-Aug-2014.txt"
gzip expression_mRNA_17-Aug-2014.txt
cortex_celltypes = (pd.read_csv('/project2/mstephens/aksarkar/projects/singlecell-ideas/data/zeisel-2015/expression_mRNA_17-Aug-2014.txt.gz', sep='\t', header=None, index_col=0, nrows=10) .reset_index(drop=True) .set_index(1))
plt.clf() fig, ax = plt.subplots(3, 3) fig.set_size_inches(6, 6) for a, k in zip(ax.ravel(), set(cortex_celltypes.loc['level1class'])): x = cortex_counts.loc[(cortex_celltypes.loc['level1class'] == k).values, 'Gpm6a'] a.hist(x, bins=np.arange(x.max() + 1), color='k') a.set_title(k) for a in ax: a[0].set_ylabel('Num cells') for a in ax.T: a[-1].set_xlabel('Num mols') fig.tight_layout()
plt.clf() fig, ax = plt.subplots(3, 3) fig.set_size_inches(6, 6) for a, k in zip(ax.ravel(), set(cortex_celltypes.loc['level1class'])): x = cortex_counts.loc[(cortex_celltypes.loc['level1class'] == k).values, 'Scn2a1'] a.hist(x, bins=np.arange(x.max() + 1), color='k') a.set_title(k) for a in ax: a[0].set_ylabel('Num cells') for a in ax.T: a[-1].set_xlabel('Num mols') fig.tight_layout()
PBMCs
Read the PBMC data.
pbmcs_68k_counts = scmodes.dataset.read_10x('/project2/mstephens/aksarkar/projects/singlecell-ideas/data/10xgenomics/fresh_68k_pbmc_donor_a/filtered_matrices_mex/hg19/', return_df=True)
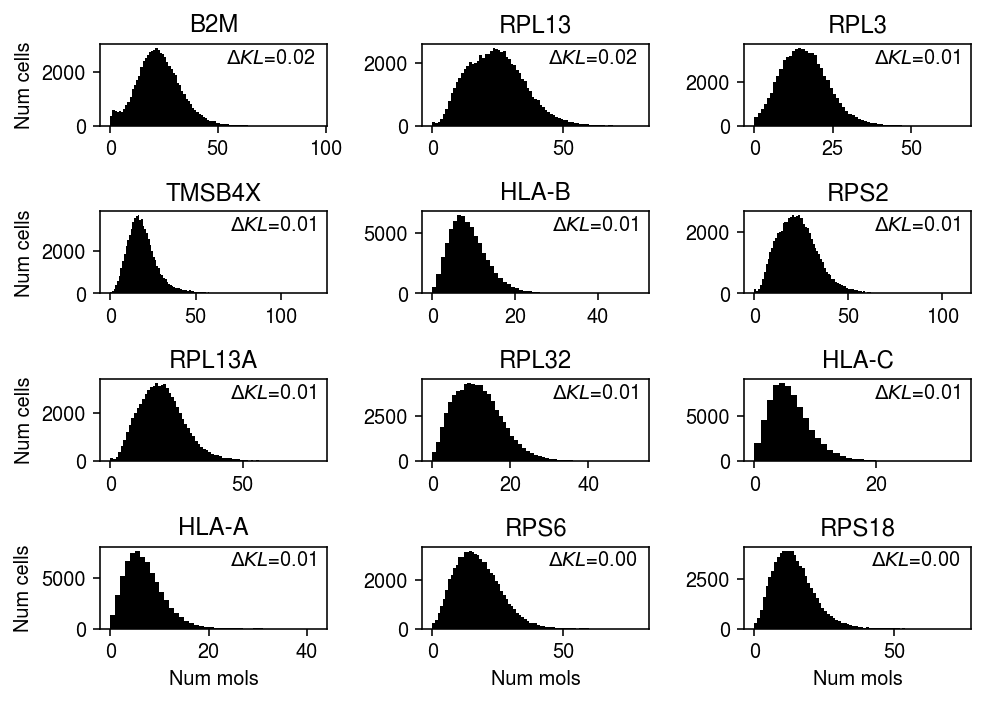
llik_diff = benchmark['pbmcs_68k']['npmle'] - benchmark['pbmcs_68k']['unimodal'] query = benchmark['pbmcs_68k'].loc[llik_diff[llik_diff > 0].sort_values(ascending=False).head(n=12).index].index plt.clf() fig, ax = plt.subplots(4, 3) fig.set_size_inches(7, 5) for a, k in zip(ax.ravel(), query): a.hist(pbmcs_68k_counts.loc[:,k], bins=np.arange(pbmcs_68k_counts.loc[:,k].max() + 1), color='k') a.text(x=.95, y=.95, s=f"$\Delta KL$={llik_diff.loc[k]:.2f}", horizontalalignment='right', verticalalignment='top', transform=a.transAxes) a.set_title(gene_info.loc[k, 'name'].upper()) for y in range(ax.shape[0]): ax[y][0].set_ylabel('Num cells') for x in range(ax.shape[1]): ax[-1][x].set_xlabel('Num mols') fig.tight_layout()